Evaluating short-term forecasting of COVID-19 cases among different epidemiological models under a Bayesian framework
Background
Forecasting of COVID-19 cases daily and weekly has been one of the challenges posed to governments and the health sector globally. To facilitate informed public health decisions, the concerned parties rely on short-term daily projections generated via predictive modeling. We calibrate stochastic variants of growth models and the standard susceptible-infectious-removed model into 1 Bayesian framework to evaluate and compare their short-term forecasts.
Results
We implement rolling-origin cross-validation to compare the short-term forecasting performance of the stochastic epidemiological models and an autoregressive moving average model across 20 countries that had the most confirmed COVID-19 cases as of August 22, 2020.
Conclusion
None of the models proved to be a gold standard across all regions, while all outperformed the autoregressive moving average model in terms of the accuracy of forecast and interpretability.
MB-GAN: Microbiome Simulation via Generative Adversarial Network
Background
Trillions of microbes inhabit the human body and have a profound effect on human health. The recent development of metagenome-wide association studies and other quantitative analysis methods accelerate the discovery of the associations between human microbiome and diseases. To assess the strengths and limitations of these analytical tools, simulating realistic microbiome datasets is critically important. However, simulating the real microbiome data is challenging because it is difficult to model their correlation structure using explicit statistical models.
Results
To address the challenge of simulating realistic microbiome data, we designed a novel simulation framework termed MB-GAN, by using a generative adversarial network (GAN) and utilizing methodology advancements from the deep learning community. MB-GAN can automatically learn from given microbial abundances and compute simulated abundances that are indistinguishable from them. In practice, MB-GAN showed the following advantages. First, MB-GAN avoids explicit statistical modeling assumptions, and it only requires real datasets as inputs. Second, unlike the traditional GANs, MB-GAN is easily applicable and can converge efficiently.
Conclusion
By applying MB-GAN to a case-control gut microbiome study of 396 samples, we demonstrated that the simulated data and the original data had similar first-order and second-order properties, including sparsity, diversities, and taxa-taxa correlations. These advantages are suitable for further microbiome methodology development where high-fidelity microbiome data are needed.
A deep learning-based model for screening and staging pneumoconiosis
Abstract
This study aims to develop an artificial intelligence (AI)-based model to assist radiologists in pneumoconiosis screening and staging using chest radiographs. The model, based on chest radiographs, was developed using a training cohort and validated using an independent test cohort. Every image in the training and test datasets were labeled by experienced radiologists in a double-blinded fashion. The computational model started by segmenting the lung field into six subregions. Then, convolutional neural network classification model was used to predict the opacity level for each subregion respectively. Finally, the diagnosis for each subject (normal, stage I, II, or III pneumoconiosis) was determined by summarizing the subregion-based prediction results. For the independent test cohort, pneumoconiosis screening accuracy was 0.973, with both sensitivity and specificity greater than 0.97. The accuracy for pneumoconiosis staging was 0.927, better than that achieved by two groups of radiologists (0.87 and 0.84, respectively). This study develops a deep learning-based model for screening and staging of pneumoconiosis using man-annotated chest radiographs. The model outperformed two groups of radiologists in the accuracy of pneumoconiosis staging. This pioneer work demonstrates the feasibility and efficiency of AI-assisted radiography screening and diagnosis in occupational lung diseases.
Tumor-suppressor function of Beclin 1 in breast cancer cells requires E-cadherin
Abstract
Beclin 1, an autophagy and haploinsufficient tumor-suppressor protein, is frequently monoallelically deleted in breast and ovarian cancers. However, the precise mechanisms by which Beclin 1 inhibits tumor growth remain largely unknown. To address this question, we performed a genome-wide CRISPR/Cas9 screen in MCF7 breast cancer cells to identify genes whose loss of function reverse Beclin 1-dependent inhibition of cellular proliferation. Small guide RNAs targeting CDH1 and CTNNA1, tumor-suppressor genes that encode cadherin/catenin complex members E-cadherin and alpha-catenin, respectively, were highly enriched in the screen. CRISPR/Cas9-mediated knockout of CDH1 or CTNNA1 reversed Beclin 1-dependent suppression of breast cancer cell proliferation and anchorage-independent growth. Moreover, deletion of CDH1 or CTNNA1 inhibited the tumor-suppressor effects of Beclin 1 in breast cancer xenografts. Enforced Beclin 1 expression in MCF7 cells and tumor xenografts increased cell surface localization of E-cadherin and decreased expression of mesenchymal markers and beta-catenin/Wnt target genes. Furthermore, CRISPR/Cas9-mediated knockout of BECN1 and the autophagy class III phosphatidylinositol kinase complex 2 (PI3KC3-C2) gene, UVRAG, but not PI3KC3-C1-specific ATG14 or other autophagy genes ATG13, ATG5, or ATG7, resulted in decreased E-cadherin plasma membrane and increased cytoplasmic E-cadherin localization. Taken together, these data reveal previously unrecognized cooperation between Beclin 1 and E-cadherin-mediated tumor suppression in breast cancer cells.
Sorting nexin 5 mediates virus-induced autophagy and immunity
Abstract
Autophagy, a process of degradation that occurs via the lysosomal pathway, has an essential role in multiple aspects of immunity, including immune system development, regulation of innate and adaptive immune and inflammatory responses, selective degradation of intracellular microorganisms, and host protection against infectious diseases1,2. Autophagy is known to be induced by stimuli such as nutrient deprivation and suppression of mTOR, but little is known about how autophagosomal biogenesis is initiated in mammalian cells in response to viral infection. Here, using genome-wide short interfering RNA screens, we find that the endosomal protein sorting nexin 5 (SNX5)3,4 is essential for virus-induced, but not for basal, stress- or endosome-induced, autophagy. We show that SNX5 deletion increases cellular susceptibility to viral infection in vitro, and that Snx5 knockout in mice enhances lethality after infection with several human viruses. Mechanistically, SNX5 interacts with beclin 1 and ATG14-containing class III phosphatidylinositol-3-kinase (PI3KC3) complex 1 (PI3KC3-C1), increases the lipid kinase activity of purified PI3KC3-C1, and is required for endosomal generation of phosphatidylinositol-3-phosphate (PtdIns(3)P) and recruitment of the PtdIns(3)P-binding protein WIPI2 to virion-containing endosomes. These findings identify a context- and organelle-specific mechanism—SNX5-dependent PI3KC3-C1 activation at endosomes—for initiation of autophagy during viral infection.
eIF5B drives integrated stress response-dependent translation of PD-L1 in lung cancer
Abstract
Cancer cells express high levels of programmed death ligand 1 (PD-L1), a ligand of the programmed cell death protein 1 (PD-1) receptor on T cells, allowing tumors to suppress T cell activity. Clinical trials utilizing antibodies that disrupt the PD-1/PD-L1 checkpoint have yielded remarkable results, with anti-PD-1 immunotherapy approved as a first-line therapy for patients with lung cancer. We used CRISPR-based screening to identify regulators of PD-L1 in human lung cancer cells, revealing potent induction of PD-L1 upon disruption of heme biosynthesis. Impairment of heme production activates the integrated stress response, allowing bypass of inhibitory upstream open reading frames in the PD-L1 5′ untranslated region, resulting in enhanced PD-L1 translation and suppression of anti-tumor immunity. We demonstrate that integrated stress-response-dependent PD-L1 translation requires the translation initiation factor eIF5B. eIF5B overexpression, which is frequent in lung adenocarcinomas and associated with poor prognosis, is sufficient to induce PD-L1. These findings illuminate mechanisms of immune checkpoint activation and identify targets for therapeutic intervention.
Molecular differences across invasive lung adenocarcinoma morphological subgroups
Background
Lung adenocarcinomas (ADCs) show heterogeneous morphological patterns that are classified into five subgroups: lepidic predominant, papillary predominant, acinar predominant, micropapillary predominant and solid predominant. The morphological classification of ADCs has been reported to be associated with patient prognosis and adjuvant chemotherapy response. However, the molecular mechanisms underlying the morphology differences among different subgroups remain largely unknown.
Results
We showed that the expression of proteins and mRNAs, but not the gene mutations copy number alterations (CNA), were significantly associated with lung ADC morphological subgroups. In addition, expression of the FOXM1 gene (which is negatively associated with patient survival) likely plays an important role in the morphological differences among different subgroups. Moreover, we found that protein abundance of PD-L1 were associated with the malignancy of subgroups. These results were validated in an independent cohort.
Conclusions
This study provides insights into the molecular differences among different lung ADC morphological subgroups, which could lead to potential subgroup-specific therapies.
Spatial molecular profiling: platforms, applications and analysis tools
Abstract
Molecular profiling technologies, such as genome sequencing and proteomics, have transformed biomedical research, but most such technologies require tissue dissociation, which leads to loss of tissue morphology and spatial information. Recent developments in spatial molecular profiling technologies have enabled the comprehensive molecular characterization of cells while keeping their spatial and morphological contexts intact. Molecular profiling data generate deep characterizations of the genetic, transcriptional and proteomic events of cells, while tissue images capture the spatial locations, organizations and interactions of the cells together with their morphology features. These data, together with cell and tissue imaging data, provide unprecedented opportunities to study tissue heterogeneity and cell spatial organization. This review aims to provide an overview of these recent developments in spatial molecular profiling technologies and the corresponding computational methods developed for analyzing such data.
HARMONIES: A Hybrid Approach for Microbiome Networks Inference via Exploiting Sparsity
Abstract
The human microbiome is a collection of microorganisms. They form complex communities and collectively affect host health. Recently, the advances in next-generation sequencing technology enable the high-throughput profiling of the human microbiome. This calls for a statistical model to construct microbial networks from the microbiome sequencing count data. As microbiome count data are high-dimensional and suffer from uneven sampling depth, over-dispersion, and zero-inflation, these characteristics can bias the network estimation and require specialized analytical tools. Here we propose a general framework, HARMONIES, Hybrid Approach foR MicrobiOme Network Inferences via Exploiting Sparsity, to infer a sparse microbiome network. HARMONIES first utilizes a zero-inflated negative binomial (ZINB) distribution to model the skewness and excess zeros in the microbiome data, as well as incorporates a stochastic process prior for sample-wise normalization. This approach infers a sparse and stable network by imposing non-trivial regularizations based on the Gaussian graphical model. In comprehensive simulation studies, HARMONIES outperformed four other commonly used methods. When using published microbiome data from a colorectal cancer study, it discovered a novel community with disease-enriched bacteria. In summary, HARMONIES is a novel and useful statistical framework for microbiome network inference, and it is available at https://github.com/shuangj00/HARMONIES.
Development of a Data Model and Data Commons for Germ Cell Tumors
Abstract
Germ cell tumors (GCTs) are considered a rare disease but are the most common solid tumors in adolescents and young adults, accounting for 15% of all malignancies in this age group. The rarity of GCTs in some groups, particularly children, has impeded progress in treatment and biologic understanding. The most effective GCT research will result from the interrogation of data sets from historical and prospective trials across institutions. However, inconsistent use of terminology among groups, different sample-labeling rules, and lack of data standards have hampered researchers’ efforts in data sharing and across-study validation. To overcome the low interoperability of data and facilitate future clinical trials, we worked with the Malignant Germ Cell International Consortium (MaGIC) and developed a GCT clinical data model as a uniform standard to curate and harmonize GCT data sets. This data model will also be the standard for prospective data collection in future trials. Using the GCT data model, we developed a GCT data commons with data sets from both MaGIC and public domains as an integrated research platform. The commons supports functions, such as data query, management, sharing, visualization, and analysis of the harmonized data, as well as patient cohort discovery. This GCT data commons will facilitate future collaborative research to advance the biologic understanding and treatment of GCTs. Moreover, the framework of the GCT data model and data commons will provide insights for other rare disease research communities into developing similar collaborative research platforms.
Bayesian multiple instance regression for modeling immunogenic neoantigens
Abstract
The relationship between tumor immune responses and tumor neoantigens is one of the most fundamental and unsolved questions in tumor immunology, and is the key to understanding the inefficiency of immunotherapy observed in many cancer patients. However, the properties of neoantigens that can elicit immune responses remain unclear. This biological problem can be represented and solved under a multiple instance learning framework, which seeks to model multiple instances (neoantigens) within each bag (patient specimen) with the continuous response (T cell infiltration) observed for each bag. To this end, we develop a Bayesian multiple instance regression method, named BMIR, using a Gaussian distribution to address continuous responses and latent binary variables to model primary instances in bags. By means of such Bayesian modeling, BMIR can learn a function for predicting the bag-level responses and for identifying the primary instances within bags, as well as give access to Bayesian statistical inference, which are elusive in existing works. We demonstrate the superiority of BMIR over previously proposed optimization-based methods for multiple instance regression through simulation and real data analyses. Our method is implemented in R package entitled “BayesianMIR” and is available at https://github.com/inmybrain/BayesianMIR.
TLR9 and Beclin 1 Crosstalk Regulates Muscle AMPK Activation in Exercise
Abstract
The activation of adenosine monophosphate-activated protein kinase (AMPK) in skeletal muscle coordinates systemic metabolic responses to exercise1. Autophagy-a lysosomal degradation pathway that maintains cellular homeostasis2-is upregulated during exercise, and a core autophagy protein, beclin 1, is required for AMPK activation in skeletal muscle3. Here we describe a role for the innate immune-sensing molecule Toll-like receptor 9 (TLR9)4, and its interaction with beclin 1, in exercise-induced activation of AMPK in skeletal muscle. Mice that lack TLR9 are deficient in both exercise-induced activation of AMPK and plasma membrane localization of the GLUT4 glucose transporter in skeletal muscle, but are not deficient in autophagy. TLR9 binds beclin 1, and this interaction is increased by energy stress (glucose starvation and endurance exercise) and decreased by a BCL2 mutation3,5 that blocks the disruption of BCL2-beclin 1 binding. TLR9 regulates the assembly of the endolysosomal phosphatidylinositol 3-kinase complex (PI3KC3-C2)-which contains beclin 1 and UVRAG-in skeletal muscle during exercise, and knockout of beclin 1 or UVRAG inhibits the cellular AMPK activation induced by glucose starvation. Moreover, TLR9 functions in a muscle-autonomous fashion in ex vivo contraction-induced AMPK activation, glucose uptake and beclin 1-UVRAG complex assembly. These findings reveal a heretofore undescribed role for a Toll-like receptor in skeletal-muscle AMPK activation and glucose metabolism during exercise, as well as unexpected crosstalk between this innate immune sensor and autophagy proteins.
Interaction between the autophagy protein Beclin 1 and Na+,K+-ATPase during starvation, exercise, and ischemia
Abstract
Autosis is a distinct form of cell death that requires both autophagy genes and the Na+,K+-ATPase pump. However, the relationship between the autophagy machinery and Na+,K+-ATPase is unknown. We explored the hypothesis that Na+,K+-ATPase interacts with the autophagy protein Beclin 1 during stress and autosis-inducing conditions. Starvation increased the Beclin 1/Na+,K+-ATPase interaction in cultured cells, and this was blocked by cardiac glycosides, inhibitors of Na+,K+-ATPase. Increases in Beclin 1/Na+,K+-ATPase interaction were also observed in tissues from starved mice, livers of patients with anorexia nervosa, brains of neonatal rats subjected to cerebral hypoxia-ischemia (HI), and kidneys of mice subjected to renal ischemia/reperfusion injury (IRI). Cardiac glycosides blocked the increased Beclin 1/Na+,K+-ATPase interaction during cerebral HI injury and renal IRI. In the mouse renal IRI model, cardiac glycosides reduced numbers of autotic cells in the kidney and improved clinical outcome. Moreover, blockade of endogenous cardiac glycosides increased Beclin 1/Na+,K+-ATPase interaction and autotic cell death in mouse hearts during exercise. Thus, Beclin 1/Na+,K+-ATPase interaction is increased in stress conditions, and cardiac glycosides decrease this interaction and autosis in both pathophysiological and physiological settings. This crosstalk between cellular machinery that generates and consumes energy during stress may represent a fundamental homeostatic mechanism.
Computational Staining of Pathology Images to Study the Tumor Microenvironment in Lung Cancer
Abstract
The spatial organization of different types of cells in tumor tissues reveals important information about the tumor microenvironment (TME). To facilitate the study of cellular spatial organization and interactions, we developed Histology-based Digital-Staining, a deep learning-based computation model, to segment the nuclei of tumor, stroma, lymphocyte, macrophage, karyorrhexis, and red blood cells from standard hematoxylin and eosin-stained pathology images in lung adenocarcinoma. Using this tool, we identified and classified cell nuclei and extracted 48 cell spatial organization-related features that characterize the TME. Using these features, we developed a prognostic model from the National Lung Screening Trial dataset, and independently validated the model in The Cancer Genome Atlas lung adenocarcinoma dataset, in which the predicted high-risk group showed significantly worse survival than the low-risk group (P = 0.001), with a HR of 2.23 (1.37-3.65) after adjusting for clinical variables. Furthermore, the image-derived TME features significantly correlated with the gene expression of biological pathways. For example, transcriptional activation of both the T-cell receptor and programmed cell death protein 1 pathways positively correlated with the density of detected lymphocytes in tumor tissues, while expression of the extracellular matrix organization pathway positively correlated with the density of stromal cells. In summary, we demonstrate that the spatial organization of different cell types is predictive of patient survival and associated with the gene expression of biological pathways. SIGNIFICANCE: These findings present a deep learning-based analysis tool to study the TME in pathology images and demonstrate that the cell spatial organization is predictive of patient survival and is associated with gene expression.See related commentary by Rodriguez-Antolin, p. 1912.
Mice With Increased Numbers of Polyploid Hepatocytes Maintain Regenerative Capacity But Develop Fewer Hepatocellular Carcinomas Following Chronic Liver Injury
Abstract
Background & aims
Thirty to 90% of hepatocytes contain whole-genome duplications, but little is known about the fates or functions of these polyploid cells or how they affect development of liver disease. We investigated the effects of continuous proliferative pressure, observed in chronically damaged liver tissues, on polyploid cells.
Methods
We studied Rosa-rtTa mice (controls) and Rosa-rtTa;TRE-short hairpin RNA mice, which have reversible knockdown of anillin, actin binding protein (ANLN). Transient administration of doxycycline increases the frequency and degree of hepatocyte polyploidy without permanently altering levels of ANLN. Mice were then given diethylnitrosamine and carbon tetrachloride (CCl4) to induce mutations, chronic liver damage, and carcinogenesis. We performed partial hepatectomies to test liver regeneration and then RNA-sequencing to identify changes in gene expression. Lineage tracing was used to rule out repopulation from non-hepatocyte sources. We imaged dividing hepatocytes to estimate the frequency of mitotic errors during regeneration. We also performed whole-exome sequencing of 54 liver nodules from patients with cirrhosis to quantify aneuploidy, a possible outcome of polyploid cell divisions.
Results
Liver tissues from control mice given CCl4 had significant increases in ploidy compared with livers from uninjured mice. Mice with knockdown of ANLN had hepatocyte ploidy above physiologic levels and developed significantly fewer liver tumors after administration of diethylnitrosamine and CCl4 compared with control mice. Increased hepatocyte polyploidy was not associated with altered regenerative capacity or tissue fitness, changes in gene expression, or more mitotic errors. Based on lineage-tracing experiments, non-hepatocytes did not contribute to liver regeneration in mice with increased polyploidy. Despite an equivalent rate of mitosis in hepatocytes of differing ploidies, we found no lagging chromosomes or micronuclei in mitotic polyploid cells. In nodules of human cirrhotic liver tissue, there was no evidence of chromosome-level copy number variations.
Conclusions
Mice with increased polyploid hepatocytes develop fewer liver tumors following chronic liver damage. Remarkably, polyploid hepatocytes maintain the ability to regenerate liver tissues during chronic damage without generating mitotic errors, and aneuploidy is not commonly observed in cirrhotic livers. Strategies to increase numbers of polypoid hepatocytes might be effective in preventing liver cancer.
Keywords
Cell Division; DEN; HCC; Mouse Model.
Dysfunctional Adaptive Immune Response in Adolescents and Young Adults With Suicide Behavior
Abstract
Background
Immune system dysfunction has been implicated in the pathophysiology of suicide behavior. Here, we conducted an exploratory analysis of immune profile differences of three groups of adolescents and young adults (ages 10-25 years): healthy controls (n = 39), at risk of major depressive disorder (MDD; at-risk, n = 33), and MDD with recent suicide behavior/ ideation (suicide behavior, n = 37).
Methods
Plasma samples were assayed for chemokines and cytokines using Bio-Plex Pro Human Chemokine 40-plex assay. Log-transformed cytokine and chemokine levels were compared after controlling for age, gender, body mass index, race, ethnicity, and C-reactive protein (CRP) levels. In post-hoc analyses to understand the effect of dysregulated immune markers identified in this exploratory analysis, their association with autoantibodies was tested in an unrelated sample (n = 166).
Results
Only levels of interleukin 4 (IL-4) differed significantly among the three groups [false discovery rate (FDR) adjusted p = 0.0007]. Participants with suicide behavior had lower IL-4 [median = 16.8 pg/ml, interquartile range (IQR) = 7.9] levels than healthy controls (median = 29.1 pg/ml, IQR = 16.1, effect size [ES] = 1.30) and those at-risk (median = 24.4 pg/ml, IQR = 16.3, ES = 1.03). IL-4 levels were negatively correlated with depression severity (r= -0.38, p = 0.024). In an unrelated sample of outpatients with MDD, levels of IL-4 were negatively correlated (all FDR p < 0.05) with several autoantibodies [54/117 in total and 12/18 against innate immune markers].
Conclusions
Adolescent and young adult patients with recent suicide behavior exhibit lower IL-4 levels. One biological consequence of reduced IL-4 levels may be increased risk of autoimmunity.
Keywords
Adaptive immunity; Autoimmunity; Depression; IL-4; Suicidality; Suicide behavior; Type 2 immune response.
VAMPr: VAriant Mapping and Prediction of Antibiotic Resistance via Explainable Features and Machine Learning
Abstract
Antimicrobial resistance (AMR) is an increasing threat to public health. Current methods of determining AMR rely on inefficient phenotypic approaches, and there remains incomplete understanding of AMR mechanisms for many pathogen-antimicrobial combinations. Given the rapid, ongoing increase in availability of high-density genomic data for a diverse array of bacteria, development of algorithms that could utilize genomic information to predict phenotype could both be useful clinically and assist with discovery of heretofore unrecognized AMR pathways. To facilitate understanding of the connections between DNA variation and phenotypic AMR, we developed a new bioinformatics tool, variant mapping and prediction of antibiotic resistance (VAMPr), to (1) derive gene ortholog-based sequence features for protein variants; (2) interrogate these explainable gene-level variants for their known or novel associations with AMR; and (3) build accurate models to predict AMR based on whole genome sequencing data. We curated the publicly available sequencing data for 3,393 bacterial isolates from 9 species that contained AMR phenotypes for 29 antibiotics. We detected 14,615 variant genotypes and built 93 association and prediction models. The association models confirmed known genetic antibiotic resistance mechanisms, such as blaKPC and carbapenem resistance consistent with the accurate nature of our approach. The prediction models achieved high accuracies (mean accuracy of 91.1% for all antibiotic-pathogen combinations) internally through nested cross validation and were also validated using external clinical datasets. The VAMPr variant detection method, association and prediction models will be valuable tools for AMR research for basic scientists with potential for clinical applicability.
Integrating Germline and Somatic Genetics to Identify Genes Associated With Lung Cancer
Abstract
Genome-wide association studies (GWAS) have successfully identified many genetic variants associated with complex traits. However, GWAS experience power issues, resulting in the failure to detect certain associated variants. Additionally, GWAS are often unable to parse the biological mechanisms of driving associations. An existing gene-based association test framework, Transcriptome-Wide Association Studies (TWAS), leverages expression quantitative trait loci data to increase the power of association tests and illuminate the biological mechanisms by which genetic variants modulate complex traits. We extend the TWAS methodology to incorporate somatic information from tumors. By integrating germline and somatic data we are able to leverage information from the nuanced somatic landscape of tumors. Thus we can augment the power of TWAS-type tests to detect germline genetic variants associated with cancer phenotypes. We use somatic and germline data on lung adenocarcinomas from The Cancer Genome Atlas in conjunction with a meta-analyzed lung cancer GWAS to identify novel genes associated with lung cancer.
Dysfunctional adaptive immune response in adolescents and young adults with suicide behavior
Abstract
Background
Immune system dysfunction has been implicated in the pathophysiology of suicide behavior. Here, we conducted an exploratory analysis of immune profile differences of three groups of adolescents and young adults (ages 10-25 years): healthy controls (n = 39), at risk of major depressive disorder (MDD; at-risk, n = 33), and MDD with recent suicide behavior/ ideation (suicide behavior, n = 37).
Methods
Plasma samples were assayed for chemokines and cytokines using Bio-Plex Pro Human Chemokine 40-plex assay. Log-transformed cytokine and chemokine levels were compared after controlling for age, gender, body mass index, race, ethnicity, and C-reactive protein (CRP) levels. In post-hoc analyses to understand the effect of dysregulated immune markers identified in this exploratory analysis, their association with autoantibodies was tested in an unrelated sample (n = 166).
Results
Only levels of interleukin 4 (IL-4) differed significantly among the three groups [false discovery rate (FDR) adjusted p = 0.0007]. Participants with suicide behavior had lower IL-4 [median = 16.8 pg/ml, interquartile range (IQR) = 7.9] levels than healthy controls (median = 29.1 pg/ml, IQR = 16.1, effect size [ES] = 1.30) and those at-risk (median = 24.4 pg/ml, IQR = 16.3, ES = 1.03). IL-4 levels were negatively correlated with depression severity (r= -0.38, p = 0.024). In an unrelated sample of outpatients with MDD, levels of IL-4 were negatively correlated (all FDR p < 0.05) with several autoantibodies [54/117 in total and 12/18 against innate immune markers].
Conclusions
Adolescent and young adult patients with recent suicide behavior exhibit lower IL-4 levels. One biological consequence of reduced IL-4 levels may be increased risk of autoimmunity.
Keywords
Adaptive immunity; Autoimmunity; Depression; IL-4; Suicidality; Suicide behavior; Type 2 immune response.
Large-Scale Profiling of RBP-circRNA Interactions From Public CLIP-Seq Datasets
Abstract
Circular RNAs are a special type of RNA that has recently attracted a lot of research interest in studying its formation and function. RNA binding proteins (RBPs) that bind circRNAs are important in these processes, but have been relatively less studied. CLIP-Seq technology has been invented and applied to profile RBP-RNA interactions on the genome-wide scale. While mRNAs are usually the focus of CLIP-Seq experiments, RBP-circRNA interactions could also be identified through specialized analysis of CLIP-Seq datasets. However, many technical difficulties are involved in this process, such as the usually short read length of CLIP-Seq reads. In this study, we created a pipeline called Clirc specialized for profiling circRNAs in CLIP-Seq data and analyzing the characteristics of RBP-circRNA interactions. In conclusion, to our knowledge, this is one of the first studies to investigate circRNAs and their binding partners through repurposing CLIP-Seq datasets, and we hope our work will become a valuable resource for future studies into the biogenesis and function of circRNAs.
Correction: LCE: An Open Web Portal to Explore Gene Expression and Clinical Associations in Lung Cancer
Abstract
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
Bayesian Multiple Instance Regression for Modeling Immunogenic Neoantigens
Abstract
The relationship between tumor immune responses and tumor neoantigens is one of the most fundamental and unsolved questions in tumor immunology, and is the key to understanding the inefficiency of immunotherapy observed in many cancer patients. However, the properties of neoantigens that can elicit immune responses remain unclear. This biological problem can be represented and solved under a multiple instance learning framework, which seeks to model multiple instances (neoantigens) within each bag (patient specimen) with the continuous response (T cell infiltration) observed for each bag. To this end, we develop a Bayesian multiple instance regression method, named BMIR, using a Gaussian distribution to address continuous responses and latent binary variables to model primary instances in bags. By means of such Bayesian modeling, BMIR can learn a function for predicting the bag-level responses and for identifying the primary instances within bags, as well as give access to Bayesian statistical inference, which are elusive in existing works. We demonstrate the superiority of BMIR over previously proposed optimization-based methods for multiple instance regression through simulation and real data analyses. Our method is implemented in R package entitled "BayesianMIR" and is available at https://github.com/inmybrain/BayesianMIR .
ConvPath: A Software Tool for Lung Adenocarcinoma Digital Pathological Image Analysis Aided by Convolutional Neural Network.
Abstract
Background
The spatial distributions of different types of cells could reveal a cancer cell's growth pattern, its relationships with the tumor microenvironment and the immune response of the body, all of which represent key "hallmarks of cancer". However, the process by which pathologists manually recognize and localize all the cells in pathology slides is extremely labor intensive and error prone.
Methods
In this study, we developed an automated cell type classification pipeline, ConvPath, which includes nuclei segmentation, convolutional neural network-based tumor cell, stromal cell, and lymphocyte classification, and extraction of tumor microenvironment-related features for lung cancer pathology images. To facilitate users in leveraging this pipeline for their research, all source scripts for ConvPath software are available at https://qbrc.swmed.edu/projects/cnn/.
Findings
The overall classification accuracy was 92.9% and 90.1% in training and independent testing datasets, respectively. By identifying cells and classifying cell types, this pipeline can convert a pathology image into a "spatial map" of tumor, stromal and lymphocyte cells. From this spatial map, we can extract features that characterize the tumor micro-environment. Based on these features, we developed an image feature-based prognostic model and validated the model in two independent cohorts. The predicted risk group serves as an independent prognostic factor, after adjusting for clinical variables that include age, gender, smoking status, and stage.
Interpretation
The analysis pipeline developed in this study could convert the pathology image into a "spatial map" of tumor cells, stromal cells and lymphocytes. This could greatly facilitate and empower comprehensive analysis of the spatial organization of cells, as well as their roles in tumor progression and metastasis.
Keywords
Cell distribution and interaction; Convolutional neural network; Deep learning; Lung adenocarcinoma; Pathology image; Prognosis.
Artificial Intelligence in Lung Cancer Pathology Image Analysis
Abstract
Objective
Accurate diagnosis and prognosis are essential in lung cancer treatment selection and planning. With the rapid advance of medical imaging technology, whole slide imaging (WSI) in pathology is becoming a routine clinical procedure. An interplay of needs and challenges exists for computer-aided diagnosis based on accurate and efficient analysis of pathology images. Recently, artificial intelligence, especially deep learning, has shown great potential in pathology image analysis tasks such as tumor region identification, prognosis prediction, tumor microenvironment characterization, and metastasis detection.
Materials and methods
In this review, we aim to provide an overview of current and potential applications for AI methods in pathology image analysis, with an emphasis on lung cancer.
Results
We outlined the current challenges and opportunities in lung cancer pathology image analysis, discussed the recent deep learning developments that could potentially impact digital pathology in lung cancer, and summarized the existing applications of deep learning algorithms in lung cancer diagnosis and prognosis.
Discussion and conclusion
With the advance of technology, digital pathology could have great potential impacts in lung cancer patient care. We point out some promising future directions for lung cancer pathology image analysis, including multi-task learning, transfer learning, and model interpretation.
Keywords
computer-aided diagnosis; deep learning; digital pathology; lung cancer; pathology image; whole-slide imaging.
Pathology Image Analysis Using Segmentation Deep Learning Algorithms
Abstract
With the rapid development of image scanning techniques and visualization software, whole slide imaging (WSI) is becoming a routine diagnostic method. Accelerating clinical diagnosis from pathology images and automating image analysis efficiently and accurately remain significant challenges. Recently, deep learning algorithms have shown great promise in pathology image analysis, such as in tumor region identification, metastasis detection, and patient prognosis. Many machine learning algorithms, including convolutional neural networks, have been proposed to automatically segment pathology images. Among these algorithms, segmentation deep learning algorithms such as fully convolutional networks stand out for their accuracy, computational efficiency, and generalizability. Thus, deep learning-based pathology image segmentation has become an important tool in WSI analysis. In this review, the pathology image segmentation process using deep learning algorithms is described in detail. The goals are to provide quick guidance for implementing deep learning into pathology image analysis and to provide some potential ways of further improving segmentation performance. Although there have been previous reviews on using machine learning methods in digital pathology image analysis, this is the first in-depth review of the applications of deep learning algorithms for segmentation in WSI analysis.
Metabolic Diversity in Human Non-Small Cell Lung Cancer Cells
Abstract
Intermediary metabolism in cancer cells is regulated by diverse cell-autonomous processes, including signal transduction and gene expression patterns, arising from specific oncogenotypes and cell lineages. Although it is well established that metabolic reprogramming is a hallmark of cancer, we lack a full view of the diversity of metabolic programs in cancer cells and an unbiased assessment of the associations between metabolic pathway preferences and other cell-autonomous processes. Here, we quantified metabolic features, mostly from the 13C enrichment of molecules from central carbon metabolism, in over 80 non-small cell lung cancer (NSCLC) cell lines cultured under identical conditions. Because these cell lines were extensively annotated for oncogenotype, gene expression, protein expression, and therapeutic sensitivity, the resulting database enables the user to uncover new relationships between metabolism and these orthogonal processes.
Systematic Analysis of Gene Expression in Lung Adenocarcinoma and Squamous Cell Carcinoma With a Case Study of FAM83A and FAM83B
Abstract
Introduction
In our previous study, we constructed a Lung Cancer Explorer (LCE) database housing lung cancer-specific expression data and clinical data from over 6700 patients in 56 studies.
Methods
Using this dataset of the largest collection of lung cancer gene expression along with our meta-analysis method, we systematically interrogated the association between gene expression and overall survival as well as the expression difference between tumor and normal (adjacent non-malignant tissue) samples in lung adenocarcinoma (ADC) and lung squamous cell carcinoma (SQCC). A case study for FAM83A and FAM83B was performed as a demonstration for hypothesis testing with our database.
Results
We showed that the reproducibility of results across studies varied by histological subtype and analysis type. Genes and pathways unique or common to the two histological subtypes were identified and the results were integrated into LCE to facilitate user exploration. In our case study, we verified the findings from a previous study on FAM83A and FAM83B in non-small cell lung cancer.
Conclusions
This study used gene expression data from a large cohort of patients to explore the molecular differences between lung ADC and SQCC.
Keywords
FAM83; gene expression difference between tumor and normal; lung cancer; meta-analysis; survival association analysis; systematic analysis.
Type and case volume of health care facility influences survival and surgery selection in cases with early‐stage non–small cell lung cancer
Abstract
Background
With the expansion of non-small cell lung cancer (NSCLC) screening methods, the percentage of cases with early-stage NSCLC is anticipated to increase. Yet it remains unclear how the type and case volume of the health care facility at which treatment occurs may affect surgery selection and overall survival for cases with early-stage NSCLC.
Methods
A total of 332,175 cases with the American Joint Committee on Cancer (AJCC) TNM stage I and stage II NSCLC who were reported to the National Cancer Data Base (NCDB) by 1302 facilities were studied. Facility type was characterized in the NCDB as community cancer program (CCP), comprehensive community cancer program (CCCP), academic/research program (ARP), or integrated network cancer program (INCP). Each facility type was dichotomized further into high-volume or low-volume groups based on the case volume. Multivariate Cox proportional hazard models, the logistic regression model, and propensity score matching were used to evaluate differences in survival and surgery selection among facilities according to type and volume.
Results
Cases from ARPs were found to have the longest survival (median, 16.4 months) and highest surgery rate (74.8%), whereas those from CCPs had the shortest survival (median, 9.7 months) and the lowest surgery rate (60.8%). The difference persisted when adjusted by potential confounders. For cases treated at CCPs, CCCPs, and ARPs, high-volume facilities had better survival outcomes than low-volume facilities. In facilities with better survival outcomes, surgery was performed for a greater percentage of cases compared with facilities with worse outcomes.
Conclusions
For cases with early-stage NSCLC, both facility type and case volume influence surgery selection and clinical outcome. Higher surgery rates are observed in facilities with better survival outcomes.
Keywords
facility type; facility volume; lung cancer; prognosis; surgery selection.
Development and Validation of a Nomogram Prognostic Model for Patients With Advanced Non-Small-Cell Lung Cancer
Abstract
Importance
Nomogram prognostic models can facilitate cancer patient treatment plans and patient enrollment in clinical trials.
Objective
The primary objective is to provide an updated and accurate prognostic model for predicting the survival of advanced non-small-cell lung cancer (NSCLC) patients, and the secondary objective is to validate a published nomogram prognostic model for NSCLC using an independent patient cohort.
Design
1817 patients with advanced NSCLC from the control arms of 4 Phase III randomized clinical trials were included in this study. Data from 524 NSCLC patients from one of these trials were used to validate a previously published nomogram and then used to develop an updated nomogram. Patients from the other 3 trials were used as independent validation cohorts of the new nomogram. The prognostic performances were comprehensively evaluated using hazard ratios, integrated area under the curve (AUC), concordance index, and calibration plots.
Setting
General community.
Main outcome
A nomogram model was developed to predict overall survival in NSCLC patients.
Results
We demonstrated the prognostic power of the previously published model in an independent cohort. The updated prognostic model contains the following variables: sex, histology, performance status, liver metastasis, hemoglobin level, white blood cell counts, peritoneal metastasis, skin metastasis, and lymphocyte percentage. This model was validated using various evaluation criteria on the 3 independent cohorts with heterogeneous NSCLC populations. In the SUN1087 patient cohort, the continuous risk score output by the nomogram achieved an integrated area under the receiver operating characteristics (ROC) curve of 0.83, a log-rank P-value of 3.87e-11, and a concordance index of 0.717. In the SAVEONCO patient cohort, the integrated area under the ROC curve was 0.755, the log-rank P-value was 4.94e-6 and the concordance index was 0.678. In the VITAL patient cohort, the integrated area under the ROC curve was 0.723, the log-rank P-value was 1.36e-11, and the concordance index was 0.654. We implemented the proposed nomogram and several previously published prognostic models on an online Web server for easy user access.
Conclusions
This nomogram model based on basic clinical features and routine lab testing predicts individual survival probabilities for advanced NSCLC and exhibits cross-study robustness.
Keywords
clinical trial data sharing; nomogram; non-small-cell lung cancer.
LCE: an open web portal to explore gene expression and clinical associations in lung cancer
Abstract
We constructed a lung cancer-specific database housing expression data and clinical data from over 6700 patients in 56 studies. Expression data from 23 genome-wide platforms were carefully processed and quality controlled, whereas clinical data were standardized and rigorously curated. Empowered by this lung cancer database, we created an open access web resource-the Lung Cancer Explorer (LCE), which enables researchers and clinicians to explore these data and perform analyses. Users can perform meta-analyses on LCE to gain a quick overview of the results on tumor vs non-malignant tissue (normal) differential gene expression and expression-survival association. Individual dataset-based survival analysis, comparative analysis, and correlation analysis are also provided with flexible options to allow for customized analyses from the user.
Development and Validation of a Pathology Image Analysis-based Predictive Model for Lung Adenocarcinoma Prognosis - A Multi-cohort Study
Abstract
Prediction of disease prognosis is essential for improving cancer patient care. Previously, we have demonstrated the feasibility of using quantitative morphological features of tumor pathology images to predict the prognosis of lung cancer patients in a single cohort. In this study, we developed and validated a pathology image-based predictive model for the prognosis of lung adenocarcinoma (ADC) patients across multiple independent cohorts. Using quantitative pathology image analysis, we extracted morphological features from H&E stained sections of formalin fixed paraffin embedded (FFPE) tumor tissues. A prediction model for patient prognosis was developed using tumor tissue pathology images from a cohort of 91 stage I lung ADC patients from the Chinese Academy of Medical Sciences (CAMS), and validated in ADC patients from the National Lung Screening Trial (NLST), and the UT Special Program of Research Excellence (SPORE) cohort. The morphological features that are associated with patient survival in the training dataset from the CAMS cohort were used to develop a prognostic model, which was independently validated in both the NLST (n = 185) and the SPORE (n = 111) cohorts. The association between predicted risk and overall survival was significant for both the NLST (Hazard Ratio (HR) = 2.20, pv = 0.01) and the SPORE cohorts (HR = 2.15 and pv = 0.044), respectively, after adjusting for key clinical variables. Furthermore, the model also predicted the prognosis of patients with stage I ADC in both the NLST (n = 123, pv = 0.0089) and SPORE (n = 68, pv = 0.032) cohorts. The results indicate that the pathology image-based model predicts the prognosis of ADC patients across independent cohorts.
A Bayesian hidden Potts mixture model for analyzing lung cancer pathology images
Abstract
Digital pathology imaging of tumor tissues, which captures histological details in high resolution, is fast becoming a routine clinical procedure. Recent developments in deep-learning methods have enabled the identification, characterization, and classification of individual cells from pathology images analysis at a large scale. This creates new opportunities to study the spatial patterns of and interactions among different types of cells. Reliable statistical approaches to modeling such spatial patterns and interactions can provide insight into tumor progression and shed light on the biological mechanisms of cancer. In this article, we consider the problem of modeling a pathology image with irregular locations of three different types of cells: lymphocyte, stromal, and tumor cells. We propose a novel Bayesian hierarchical model, which incorporates a hidden Potts model to project the irregularly distributed cells to a square lattice and a Markov random field prior model to identify regions in a heterogeneous pathology image. The model allows us to quantify the interactions between different types of cells, some of which are clinically meaningful. We use Markov chain Monte Carlo sampling techniques, combined with a double Metropolis-Hastings algorithm, in order to simulate samples approximately from a distribution with an intractable normalizing constant. The proposed model was applied to the pathology images of $205$ lung cancer patients from the National Lung Screening trial, and the results show that the interaction strength between tumor and stromal cells predicts patient prognosis (P = $0.005$). This statistical methodology provides a new perspective for understanding the role of cell-cell interactions in cancer progression.
Validation of the 12-gene Predictive Signature for Adjuvant Chemotherapy Response in Lung Cancer
Abstract
Purpose
Response to adjuvant chemotherapy after tumor resection varies widely among patients with non-small cell lung cancer (NSCLC); therefore, it is of clinical importance to prospectively predict who will benefit from adjuvant chemotherapy before starting the treatment. The goal of this study is to validate a 12-gene adjuvant chemotherapy predictive signature developed from a previous study using a clinical-grade assay.
Experimental design
We developed a clinical-grade assay for formalin-fixed, paraffin-embedded (FFPE) samples using the NanoString nCounter platform to measure the mRNA expression of the previously published 12-gene set. The predictive performance was validated in a cohort of 207 patients with early-stage resected NSCLC with matched propensity score of adjuvant chemotherapy.
Results
The effects of adjuvant chemotherapy were significantly different in patients from the predicted adjuvant chemotherapy benefit group and those in the predicted adjuvant chemotherapy nonbenefit group (P = 0.0056 for interaction between predicted risk group and adjuvant chemotherapy). Specifically, in the predicted adjuvant chemotherapy benefit group, the patients receiving adjuvant chemotherapy had significant recurrence-free survival (RFS) benefit (HR = 0.34; P = 0.016; adjuvant chemotherapy vs. nonadjuvant chemotherapy), while in the predicted adjuvant chemotherapy nonbenefit group, the patients receiving adjuvant chemotherapy actually had worse RFS (HR = 1.86; P = 0.14; adjuvant chemotherapy vs. nonadjuvant chemotherapy) than those who did not receive adjuvant chemotherapy.
Conclusions
This study validated that the 12-gene signature and the FFPE-based clinical assay predict that patients whose resected lung adenocarcinomas exhibit an adjuvant chemotherapy benefit gene expression pattern and who then receive adjuvant chemotherapy have significant survival advantage compared with patients whose tumors exhibit the benefit pattern but do not receive adjuvant chemotherapy.
DIGREM: an integrated web-based platform for detecting effective multi-drug combinations
Abstract
Motivation
Synergistic drug combinations are a promising approach to achieve a desirable therapeutic effect in complex diseases through the multi-target mechanism. However, in vivo screening of all possible multi-drug combinations remains cost-prohibitive. An effective and robust computational model to predict drug synergy in silico will greatly facilitate this process.
Results
We developed DIGREM (Drug-Induced Genomic Response models for identification of Effective Multi-drug combinations), an online tool kit that can effectively predict drug synergy. DIGREM integrates DIGRE, IUPUI_CCBB, gene set-based and correlation-based models for users to predict synergistic drug combinations with dose-response information and drug-treated gene expression profiles.
Availability and implementation
http://lce.biohpc.swmed.edu/drugcombination
Supplementary information
Supplementary data are available at Bioinformatics online.
A Bayesian Zero-Inflated Negative Binomial Regression Model for the Integrative Analysis of Microbiome Data
Abstract
Microbiome omics approaches can reveal intriguing relationships between the human microbiome and certain disease states. Along with identification of specific bacteria taxa associated with diseases, recent scientific advancements provide mounting evidence that metabolism, genetics, and environmental factors can all modulate these microbial effects. However, the current methods for integrating microbiome data and other covariates are severely lacking. Hence, we present an integrative Bayesian zero-inflated negative binomial regression model that can both distinguish differentially abundant taxa with distinct phenotypes and quantify covariate-taxa effects. Our model demonstrates good performance using simulated data. Furthermore, we successfully integrated microbiome taxonomies and metabolomics in two real microbiome datasets to provide biologically interpretable findings. In all, we proposed a novel integrative Bayesian regression model that features bacterial differential abundance analysis and microbiome-covariate effects quantifications, which makes it suitable for general microbiome studies.
A Comparative Study of Rank Aggregation Methods for Partial and Top Ranked Lists in Genomic Applications
Abstract
Rank aggregation (RA), the process of combining multiple ranked lists into a single ranking, has played an important role in integrating information from individual genomic studies that address the same biological question. In previous research, attention has been focused on aggregating full lists. However, partial and/or top ranked lists are prevalent because of the great heterogeneity of genomic studies and limited resources for follow-up investigation. To be able to handle such lists, some ad hoc adjustments have been suggested in the past, but how RA methods perform on them (after the adjustments) has never been fully evaluated. In this article, a systematic framework is proposed to define different situations that may occur based on the nature of individually ranked lists. A comprehensive simulation study is conducted to examine the performance characteristics of a collection of existing RA methods that are suitable for genomic applications under various settings simulated to mimic practical situations. A non-small cell lung cancer data example is provided for further comparison. Based on our numerical results, general guidelines about which methods perform the best/worst, and under what conditions, are provided. Also, we discuss key factors that substantially affect the performance of the different methods.
GeNeCK: A Web Server for Gene Network Construction and Visualization
Abstract
Background
Reverse engineering approaches to infer gene regulatory networks using computational methods are of great importance to annotate gene functionality and identify hub genes. Although various statistical algorithms have been proposed, development of computational tools to integrate results from different methods and user-friendly online tools is still lagging.
Results
We developed a web server that efficiently constructs gene networks from expression data. It allows the user to use ten different network construction methods (such as partial correlation-, likelihood-, Bayesian- and mutual information-based methods) and integrates the resulting networks from multiple methods. Hub gene information, if available, can be incorporated to enhance performance.
Conclusions
GeNeCK is an efficient and easy-to-use web application for gene regulatory network construction. It can be accessed at http://lce.biohpc.swmed.edu/geneck .
Keywords
Bayesian; Correlation; Ensemble; Gene network; Hub gene; Likelihood; Mutual information; Statistical method; Visualization; Web server.
Development and Validation of a Nomogram Prognostic Model for SCLC Patients
Abstract
Introduction
SCLC accounts for almost 15% of lung cancer cases in the United States. Nomogram
prognostic models could greatly facilitate risk stratification and treatment
planning, as well as more refined enrollment criteria for clinical trials. We
developed and validated a new nomogram prognostic model for SCLC patients using
a large SCLC patient cohort from the National Cancer Database (NCDB).
Methods
Clinical data for 24,680 SCLC patients diagnosed from 2004 to 2011 were used to
develop the nomogram prognostic model. The model was then validated using an
independent cohort of 9700 SCLC patients diagnosed from 2012 to 2013. The
prognostic performance was evaluated using p value, concordance index and
integrated area under the (time-dependent receiver operating characteristic)
curve (AUC).
Results
The following variables were contained in the final prognostic model: age, sex,
race, ethnicity, Charlson/Deyo score, TNM stage (assigned according to the
American Joint Committee on Cancer [AJCC] eighth edition), treatment type
(combination of surgery, radiation therapy, and chemotherapy), and laterality.
The model was validated in an independent testing group with a concordance index
of 0.722 ± 0.004 and an integrated area under the curve of 0.79. The nomogram
model has a significantly higher prognostic accuracy than previously developed
models, including the AJCC eighth edition TNM-staging system. We implemented the
proposed nomogram and four previously published nomograms in an online
webserver.
Conclusions
We developed a nomogram prognostic model for SCLC patients, and validated the
model using an independent patient cohort. The nomogram performs better than
earlier models, including models using AJCC staging.
Previous
Comprehensive analysis of lung cancer pathology images to discover tumor shape features that predict survival outcome
Abstract
Pathology slide images capture tumor histomorphological details in high resolution. However, manual detection and characterization of tumor regions in pathology slides is labor intensive and subjective. Using a deep convolutional neural network (CNN), we developed an automated tumor region recognition system for lung cancer pathology slides. From the identified regions, we extracted 22 well-defined tumor shape features and found that 15 of them were significantly associated with patient survival outcome in lung adenocarcinoma patients from the National Lung Screening Trial. A tumor shape-based prognostic model was developed and validated in an independent patient cohort (n=389). The predicted high-risk group had significantly worse survival than the low-risk group (p value = 0.0029). Predicted risk group serves as an independent prognostic factor (high-risk vs. low-risk, hazard ratio = 2.25, 95% CI 1.34-3.77, p value = 0.0022) after adjusting for age, gender, smoking status, and stage. This study provides new insights into the relationship between tumor shape and patient prognosis.
Increased autophagy blocks HER2-mediated breast tumorigenesis
Abstract
Allelic loss of the autophagy gene, beclin 1/BECN1, increases the risk of patients developing aggressive, including human epidermal growth factor receptor 2 (HER2)-positive, breast cancers; however, it is not known whether autophagy induction may be beneficial in preventing HER2-positive breast tumor growth. We explored the regulation of autophagy in breast cancer cells by HER2 in vitro and the effects of genetic and pharmacological strategies to increase autophagy on HER2-driven breast cancer growth in vivo. Our findings demonstrate that HER2 interacts with Beclin 1 in breast cancer cells and inhibits autophagy. Mice with increased basal autophagy due to a genetically engineered mutation in Becn1 are protected from HER2-driven mammary tumorigenesis, and HER2 fails to inhibit autophagy in primary cells derived from these mice. Moreover, treatment of mice with HER2-positive human breast cancer xenografts with the Tat-Beclin 1 autophagy-inducing peptide inhibits tumor growth as effectively as a clinically used HER2 tyrosine kinase inhibitor (TKI). This inhibition of tumor growth is associated with a robust induction of autophagy, a disruption of HER2/Beclin 1 binding, and a transcriptional signature in the tumors distinct from that observed with HER2 TKI treatment. Taken together, these findings indicate that the HER2-mediated inhibition of Beclin 1 and autophagy likely contributes to HER2-mediated tumorigenesis and that strategies to block HER2/Beclin 1 binding and/or increase autophagy may represent a new therapeutic approach for HER2-positive breast cancers.
Microvessel prediction in H&E Stained Pathology Images using fully convolutional neural networks
Abstract
Pathological angiogenesis has been identified in many malignancies as a potential prognostic factor and target for therapy. In most cases, angiogenic analysis is based on the measurement of microvessel density (MVD) detected by immunostaining of CD31 or CD34. However, most retrievable public data is generally composed of Hematoxylin and Eosin (H&E)-stained pathology images, for which is difficult to get the corresponding immunohistochemistry images. The role of microvessels in H&E stained images has not been widely studied due to their complexity and heterogeneity. Furthermore, identifying microvessels manually for study is a labor-intensive task for pathologists, with high inter- and intra-observer variation. Therefore, it is important to develop automated microvessel-detection algorithms in H&E stained pathology images for clinical association analysis.
Main bronchus location is a predictor for metastasis and prognosis in lung adenocarcinoma: A large cohort analysis
Abstract
Objectives
In the literature, inconsistent associations between the primary locations of lung adenocarcinomas (ADCs) with patient prognosis have been reported, due to varying definitions for central and peripheral locations. In this study, we investigated the clinical characteristics and prognoses of ADCs located in the main bronchus.
Methods
A total of 397,189 lung ADCs registered from 2004 to 2013 in the National Cancer Database (NCDB) were extracted and divided into main bronchus-located ADCs (2.5%, N = 10,111) and non-main bronchus ADCs (97.5%, N = 387,078). The ADCs located in the main bronchus and those not in the main bronchus were compared in terms of patient prognosis, lymph node involvement, distant metastases and other clinical features, including rate of curative-intent resection, histologic grade, and stage.
Results
ADCs located in the main bronchus had significantly worse patient survival than those in the non-main bronchus, both for all patients (HR = 1.82, 95% CI 1.78–1.86) and for those undergoing curative-intent resection (HR = 2.49, 95% CI 2.23–2.78). Furthermore, ADCs located in the main bronchus had a significantly higher rate of lymph node involvement and distant metastasis than those not in the main bronchus, when stratified by tumor size (trend test, p < e−16). Multivariate analysis of overall survival showed that main bronchus location is a prognostic factor (HR = 1.15, 95% CI 1.08–1.23) independent of other clinical factors.
Conclusions
Main bronchus location is an independent predictor for metastasis and worse outcomes irrespective of stage and treatment. Tumor primary location might be considered in prognostication and treatment planning.
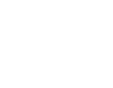
Genomic regression analysis of coordinated expression.
Abstract
Co-expression analysis is widely used to predict gene function and to identify functionally related gene sets. However, co-expression analysis using human cancer transcriptomic data is confounded by somatic copy number alterations (SCNA), which produce co-expression signatures based on physical proximity rather than biological function. To better understand gene-gene co-expression based on biological regulation but not SCNA, we describe a method termed "Genomic Regression Analysis of Coordinated Expression" (GRACE) to adjust for the effect of SCNA in co-expression analysis. The results from analyses of TCGA, CCLE, and NCI60 data sets show that GRACE can improve our understanding of how a transcriptional network is re-wired in cancer. A user-friendly web database populated with data sets from The Cancer Genome Atlas (TCGA) is provided to allow customized query.
A two-stage approach of gene network analysis for high-dimensional heterogeneous data.
Abstract
Gaussian graphical models have been widely used to construct gene regulatory networks from gene expression data. Most existing methods for Gaussian graphical models are designed to model homogeneous data, assuming a single Gaussian distribution. In practice, however, data may consist of gene expression studies with unknown confounding factors, such as study cohort, microarray platforms, experimental batches, which produce heterogeneous data, and hence lead to false positive edges or low detection power in resulting network, due to those unknown factors. To overcome this problem and improve the performance in constructing gene networks, we propose a two-stage approach to construct a gene network from heterogeneous data. The first stage is to perform a clustering analysis in order to assign samples to a few clusters where the samples in each cluster are approximately homogeneous, and the second stage is to conduct an integrative analysis of networks from each cluster. In particular, we first apply a model-based clustering method using the singular value decomposition for high-dimensional data, and then integrate the networks from each cluster using the integrative $\psi$-learning method. The proposed method is based on an equivalent measure of partial correlation coefficients in Gaussian graphical models, which is computed with a reduced conditional set and thus it is useful for high-dimensional data. We compare the proposed two-stage learning approach with some existing methods in various simulation settings, and demonstrate the robustness of the proposed method. Finally, it is applied to integrate multiple gene expression studies of lung adenocarcinoma to identify potential therapeutic targets and treatment biomarkers.
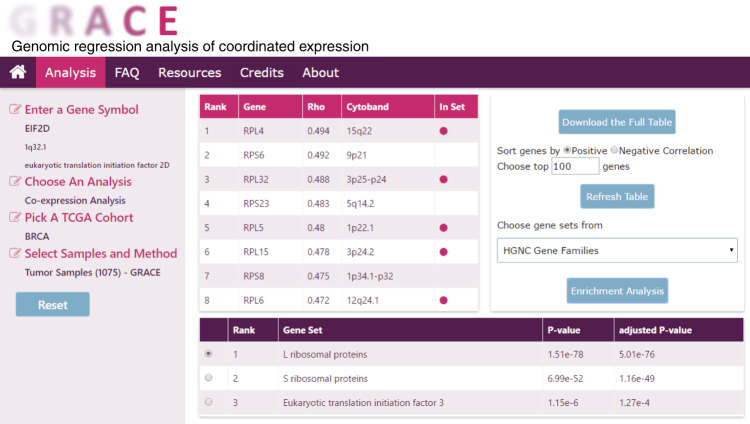
A Community Challenge for Inferring Genetic Predictors of Gene Essentialities through Analysis of a Functional Screen of Cancer Cell Lines.
Abstract
We report the results of a DREAM challenge designed to predict relative genetic essentialities based on a novel dataset testing 98,000 shRNAs against 149 molecularly characterized cancer cell lines. We analyzed the results of over 3,000 submissions over a period of 4 months. We found that algorithms combining essentiality data across multiple genes demonstrated increased accuracy; gene expression was the most informative molecular data type; the identity of the gene being predicted was far more important than the modeling strategy; well-predicted genes and selected molecular features showed enrichment in functional categories; and frequently selected expression features correlated with survival in primary tumors. This study establishes benchmarks for gene essentiality prediction, presents a community resource for future comparison with this benchmark, and provides insights into factors influencing the ability to predict gene essentiality from functional genetic screens. This study also demonstrates the value of releasing pre-publication data publicly to engage the community in an open research collaboration.
Evaluation of the 7th and 8th editions of the AJCC/UICC TNM staging systems for lung cancer in a large North American cohort.
Abstract
Purpose
The new 8th American Joint Committee on Cancer (AJCC)/International Union for Cancer Control (UICC) lung cancer staging system was developed and internally validated using the International Association for the Study of Lung Cancer (IASLC) database, but external validation is needed. The goal of this study is to validate the discriminatory ability and prognostic performance of this new staging system in a larger, independent non-small cell lung cancer (NSCLC) cohort with greater emphasis on North American patients.
Methods
A total of 858,909 NSCLC cases with one malignant primary tumor collected from 2004 to 2013 in the National Cancer Database (NCDB) were analyzed. The primary coding guidelines of the Collaborative Staging Manual and Coding Instructions for the new 8th edition AJCC/UICC lung cancer staging system was used to define the new T, M and TNM stages for all patients in the database. Kaplan-Meier curves, Cox regression models and time-dependent receiver operating characteristics were used to compare the discriminatory ability and prognostic performance of the 7th and the revised 8th T, M categories and overall stages.
Results
We demonstrated that the 8th staging system provides better discriminatory ability than the 7th staging system and predicts prognosis for NSCLC patients using the NCDB. There were significant survival differences between adjacent groups defined by both clinical staging and pathologic staging systems. These staging parameters were significantly associated with survival after adjusting for other factors.
Conclusions
The updated T, M, and overall TNM stage of the 8th staging system show improvement compared to the 7th edition in discriminatory ability between adjacent subgroups and are independent predictors for prognosis.
Abstract
Background
Clustered regularly-interspaced short palindromic repeats (CRISPR) screens are usually implemented in cultured cells to identify genes with critical functions. Although several methods have been developed or adapted to analyze CRISPR screening data, no single specific algorithm has gained popularity. Thus, rigorous procedures are needed to overcome the shortcomings of existing algorithms.
Methods
We developed a Permutation-Based Non-Parametric Analysis (PBNPA) algorithm, which computes p-values at the gene level by permuting sgRNA labels, and thus it avoids restrictive distributional assumptions. Although PBNPA is designed to analyze CRISPR data, it can also be applied to analyze genetic screens implemented with siRNAs or shRNAs and drug screens.
Results
We compared the performance of PBNPA with competing methods on simulated data as well as on real data. PBNPA outperformed recent methods designed for CRISPR screen analysis, as well as methods used for analyzing other functional genomics screens, in terms of Receiver Operating Characteristics (ROC) curves and False Discovery Rate (FDR) control for simulated data under various settings. Remarkably, the PBNPA algorithm showed better consistency and FDR control on published real data as well.
Conclusions
PBNPA yields more consistent and reliable results than its competitors, especially when the data quality is low. R package of PBNPA is available at: https://cran.r-project.org/web/packages/PBNPA/ .
Automatic extraction of cell nuclei from H&E-stained histopathological images.
Abstract
Extraction of cell nuclei from hematoxylin and eosin (H&E)-stained histopathological images is an essential preprocessing step in computerized image analysis for disease detection, diagnosis, and prognosis. We present an automated cell nuclei segmentation approach that works with H&E-stained images. A color deconvolution algorithm was first applied to the image to get the hematoxylin channel. Using a morphological operation and thresholding technique on the hematoxylin channel image, candidate target nuclei and background regions were detected, which were then used as markers for a marker-controlled watershed transform segmentation algorithm. Moreover, postprocessing was conducted to split the touching nuclei. For each segmented region from the previous steps, the regional maximum value positions were identified as potential nuclei centers. These maximum values were further grouped into [Formula: see text]-clusters, and the locations within each cluster were connected with the minimum spanning tree technique. Then, these connected positions were utilized as new markers for a watershed segmentation approach. The final number of nuclei at each region was determined by minimizing an objective function that iterated all of the possible [Formula: see text]-values. The proposed method was applied to the pathological images of the tumor tissues from The Cancer Genome Atlas study. Experimental results show that the proposed method can lead to promising results in terms of segmentation accuracy and separation of touching nuclei.
Lung Cancer Pathological Image Analysis Using a Hidden Potts Model.
Abstract
Nowadays, many biological data are acquired via images. In this article, we study the pathological images scanned from 205 patients with lung cancer with the goal to find out the relationship between the survival time and the spatial distribution of different types of cells, including lymphocyte, stroma, and tumor cells. Toward this goal, we model the spatial distribution of different types of cells using a modified Potts model for which the parameters represent interactions between different types of cells and estimate the parameters of the Potts model using the double Metropolis-Hastings algorithm. The double Metropolis-Hastings algorithm allows us to simulate samples approximately from a distribution with an intractable normalizing constant. Our numerical results indicate that the spatial interaction between the lymphocyte and tumor cells is significantly associated with the patient's survival time, and it can be used together with the cell count information to predict the survival of the patients.
Integrative gene set enrichment analysis utilizing isoform-specific expression.
Abstract
Gene set enrichment analysis (GSEA) aims at identifying essential pathways, or more generally, sets of biologically related genes that are involved in complex human diseases. In the past, many studies have shown that GSEA is a very useful bioinformatics tool that plays critical roles in the innovation of disease prevention and intervention strategies. Despite its tremendous success, it is striking that conclusions of GSEA drawn from isolated studies are often sparse, and different studies may lead to inconsistent and sometimes contradictory results. Further, in the wake of next generation sequencing technologies, it has been made possible to measure genome-wide isoform-specific expression levels, calling for innovations that can utilize the unprecedented resolution. Currently, enormous amounts of data have been created from various RNA-seq experiments. All these give rise to a pressing need for developing integrative methods that allow for explicit utilization of isoform-specific expression, to combine multiple enrichment studies, in order to enhance the power, reproducibility, and interpretability of the analysis. We develop and evaluate integrative GSEA methods, based on two-stage procedures, which, for the first time, allow statistically efficient use of isoform-specific expression from multiple RNA-seq experiments. Through simulation and real data analysis, we show that our methods can greatly improve the performance in identifying essential gene sets compared to existing methods that can only use gene-level expression.
Integrative Analysis of Gene Networks and Their Application to Lung Adenocarcinoma Studies.
Abstract
The construction of gene regulatory networks (GRNs) is an essential component of biomedical research to determine disease mechanisms and identify treatment targets. Gaussian graphical models (GGMs) have been widely used for constructing GRNs by inferring conditional dependence among a set of gene expressions. In practice, GRNs obtained by the analysis of a single data set may not be reliable due to sample limitations. Therefore, it is important to integrate multiple data sets from comparable studies to improve the construction of a GRN. In this article, we introduce an equivalent measure of partial correlation coefficients in GGMs and then extend the method to construct a GRN by combining the equivalent measures from different sources. Furthermore, we develop a method for multiple data sets with a natural missing mechanism to accommodate the differences among different platforms in multiple sources of data. Simulation results show that this integrative analysis outperforms the standard methods and can detect hub genes in the true network. The proposed integrative method was applied to 12 lung adenocarcinoma data sets collected from different studies. The constructed network is consistent with the current biological knowledge and reveals new insights about lung adenocarcinoma.
Enhanced construction of gene regulatory networks using hub gene information.
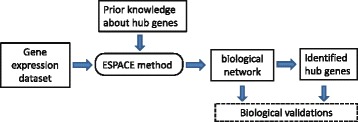
Abstract
Background
Gene regulatory networks reveal how genes work together to carry out their biological functions. Reconstructions of gene networks from gene expression data greatly facilitate our understanding of underlying biological mechanisms and provide new opportunities for biomarker and drug discoveries. In gene networks, a gene that has many interactions with other genes is called a hub gene, which usually plays an essential role in gene regulation and biological processes. In this study, we developed a method for reconstructing gene networks using a partial correlation-based approach that incorporates prior information about hub genes. Through simulation studies and two real-data examples, we compare the performance in estimating the network structures between the existing methods and the proposed method.
Results
In simulation studies, we show that the proposed strategy reduces errors in estimating network structures compared to the existing methods. When applied to Escherichia coli, the regulation network constructed by our proposed ESPACE method is more consistent with current biological knowledge than the SPACE method. Furthermore, application of the proposed method in lung cancer has identified hub genes whose mRNA expression predicts cancer progress and patient response to treatment.
Conclusions
We have demonstrated that incorporating hub gene information in estimating network structures can improve the performance of the existing methods.
Learning gene regulatory networks from next generation sequencing data.
Abstract
In recent years, next generation sequencing (NGS) has gradually replaced microarray as the major platform in measuring gene expressions. Compared to microarray, NGS has many advantages, such as less noise and higher throughput. However, the discreteness of NGS data also challenges the existing statistical methodology. In particular, there still lacks an appropriate statistical method for reconstructing gene regulatory networks using NGS data in the literature. The existing local Poisson graphical model method is not consistent and can only infer certain local structures of the network. In this article, we propose a random effect model-based transformation to continuize NGS data and then we transform the continuized data to Gaussian via a semiparametric transformation and apply an equivalent partial correlation selection method to reconstruct gene regulatory networks. The proposed method is consistent. The numerical results indicate that the proposed method can lead to much more accurate inference of gene regulatory networks than the local Poisson graphical model and other existing methods. The proposed data-continuized transformation fills the theoretical gap for how to transform discrete data to continuous data and facilitates NGS data analysis. The proposed data-continuized transformation also makes it feasible to integrate different types of data, such as microarray and RNA-seq data, in reconstruction of gene regulatory networks.

Abstract
RNA-binding proteins play important roles in the various stages of RNA maturation through binding to its target RNAs. Cross-linking immunoprecipitation coupled with high-throughput sequencing (CLIP-Seq) has made it possible to identify the targeting sites of RNA-binding proteins in various cell culture systems and tissue types on a genome-wide scale. Several Hidden Markov model-based (HMM) approaches have been suggested to identify protein-RNA binding sites from CLIP-Seq datasets. In this chapter, we describe how HMM can be applied to analyze CLIP-Seq datasets, including the bioinformatics preprocessing steps to extract count information from the sequencing data before HMM and the downstream analysis steps following peak-calling.
Comprehensive Computational Pathological Image Analysis Predicts Lung Cancer Prognosis.
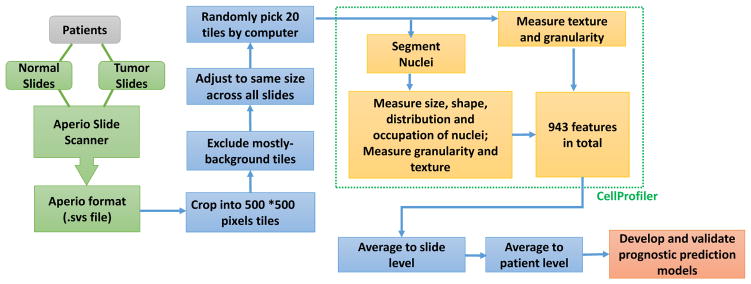
Abstract
Introduction
Pathological examination of histopathological slides is a routine clinical procedure for lung cancer diagnosis and prognosis. Although the classification of lung cancer has been updated to become more specific, only a small subset of the total morphological features are taken into consideration. The vast majority of the detailed morphological features of tumor tissues, particularly tumor cells' surrounding microenvironment, are not fully analyzed. The heterogeneity of tumor cells and close interactions between tumor cells and their microenvironments are closely related to tumor development and progression. The goal of this study is to develop morphological feature-based prediction models for the prognosis of patients with lung cancer.
Method
We developed objective and quantitative computational approaches to analyze the morphological features of pathological images for patients with NSCLC. Tissue pathological images were analyzed for 523 patients with adenocarcinoma (ADC) and 511 patients with squamous cell carcinoma (SCC) from The Cancer Genome Atlas lung cancer cohorts. The features extracted from the pathological images were used to develop statistical models that predict patients' survival outcomes in ADC and SCC, respectively.
Results
We extracted 943 morphological features from pathological images of hematoxylin and eosin-stained tissue and identified morphological features that are significantly associated with prognosis in ADC and SCC, respectively. Statistical models based on these extracted features stratified NSCLC patients into high-risk and low-risk groups. The models were developed from training sets and validated in independent testing sets: a predicted high-risk group versus a predicted low-risk group (for patients with ADC: hazard ratio = 2.34, 95% confidence interval: 1.12-4.91, p = 0.024; for patients with SCC: hazard ratio = 2.22, 95% confidence interval: 1.15-4.27, p = 0.017) after adjustment for age, sex, smoking status, and pathologic tumor stage.
Conclusions
The results suggest that the quantitative morphological features of tumor pathological images predict prognosis in patients with lung cancer.
SHOX2 is a Potent Independent Biomarker to Predict Survival of WHO Grade II-III Diffuse Gliomas.
Abstract
Background
Diffuse gliomas, grades II and III, hereafter called lower-grade gliomas (LGG), have variable, difficult to predict clinical courses, resulting in multiple studies to identify prognostic biomarkers. The purpose of this study was to assess expression or methylation of the homeobox family gene SHOX2 as independent markers for LGG survival.
Methods
We downloaded publically available glioma datasets for gene expression and methylation. The Cancer Genome Atlas (TCGA) (LGG, n=516) was used as a training set, and three other expression datasets (n=308) and three other methylation datasets (n=320), were used for validation. We performed Kaplan-Meier survival curves and univariate and multivariate Cox regression model analyses.
Findings
SHOX2 expression and gene body methylation varied among LGG patients and highly significantly predicted poor overall survival. While they were tightly correlated, SHOX2 expression appeared more potent as a prognostic marker and was used for most further studies. The SHOX2 prognostic roles were maintained after analyses by histology subtypes or tumor grade. We found that the combination of SHOX2 expression and IDH genotype status identified a subset of LGG patients with IDH wild-type (IDHwt) and low SHOX2 expression with considerably favorable survival. We further investigated the combination of SHOX2 with other known clinically relevant markers of LGG (TERT expression, 1p/19q chromosome co-deletion, MGMT methylation, ATRX mutation and NES expression). When combined with SHOX2 expression, we identified subsets of LGG patients with significantly favorable survival outcomes, especially in the subgroup with worse prognosis for each individual marker. Finally, multivariate analysis demonstrated that SHOX2 was a potent independent survival marker.
Interpretation
We have identified that SHOX2 expression or methylation are potent independent prognostic indicators for predicting LGG patient survival, and have potential to identify an important subset of LGG patients with IDHwt status with significantly better overall survival. The combination of IDH or other relevant markers with SHOX2 identified LGG subsets with significantly different survival outcomes, and further understanding of these subsets may benefit therapeutic target identification and therapy selections for glioma patients.
Extracellular Volume Overload and Increased Vasoconstriction in Patients With Recurrent Intradialytic Hypertension.
Abstract
Background
Intradialytic hypertension (IH) occurs frequently in some hemodialysis patients and increases mortality risk. We simultaneously compared pre-dialysis, post-dialysis and changes in extracellular volume and hemodynamics in recurrent IH patients and controls.
Method
We performed a case-control study among prevalent hemodialysis patients with recurrent IH and hypertensive hemodialysis controls. We used bioimpedance spectroscopy and impedance cardiography to compare pre-dialysis, post-dialysis, and intradialytic change in total body water (TBW) and extracellular water (ECW), as well as cardiac index (CI) and total peripheral resistance index (TPRI).
Results
The ECW/TBW was 0.453 (0.05) pre-dialysis and 0.427 (0.04) post-dialysis in controls vs. 0.478 (0.03) and 0.461 (0.03) in IH patients (p=0.01 post-dialysis). The ECW/TBW change was -0.027 (0.03) in controls and -0.013 (0.02) in IH patients (p=0.1). In controls, pre- and post-dialysis TPRI were 3254 (994) and 2469 (529) dynes/sec/cm2/m2 vs. 2983 (747) and 3408 (980) dynes/sec/cm2/m2 in IH patients (p=0.002 post-dialysis). There were between-group differences in TPRI change (0=0.0001), but not CI (p=0.09).
Conclusions
Recurrent intradialytic hypertension is associated with higher post-dialysis extracellular volume and TPRI. Intradialytic TPRI surges account for the vasoconstrictive state post-dialysis, but intradialytic fluid shifts may contribute to post-hemodialysis volume expansion.
Crowdsourced assessment of common genetic contribution to predicting anti-TNF treatment response in rheumatoid arthritis.
Abstract
Rheumatoid arthritis (RA) affects millions world-wide. While anti-TNF treatment is widely used to reduce disease progression, treatment fails in ∼one-third of patients. No biomarker currently exists that identifies non-responders before treatment. A rigorous community-based assessment of the utility of SNP data for predicting anti-TNF treatment efficacy in RA patients was performed in the context of a DREAM Challenge (http://www.synapse.org/RA_Challenge). An open challenge framework enabled the comparative evaluation of predictions developed by 73 research groups using the most comprehensive available data and covering a wide range of state-of-the-art modelling methodologies. Despite a significant genetic heritability estimate of treatment non-response trait (h2=0.18, P value=0.02), no significant genetic contribution to prediction accuracy is observed. Results formally confirm the expectations of the rheumatology community that SNP information does not significantly improve predictive performance relative to standard clinical traits, thereby justifying a refocusing of future efforts on collection of other data.
Comparison of Ambulatory Blood Pressure Patterns in Patients With Intradialytic Hypertension and Hemodialysis Controls.
Abstract
Background
Intradialytic hypertension (IH) patients have higher mortality risk than other hemodialysis patients and have been shown to have higher ambulatory blood pressure (BP). We hypothesized that interdialytic BP patterns would differ in IH patients and hypertensive hemodialysis controls.
Methods
We consecutively screened hemodialysis patients at our university-affiliated units. Based on pre and post-HD BP measurements during the prior 2 week period, we identified IH patients and demographically matched hemodialysis controls. We measured ambulatory interdialytic BP, flow-mediated vasodilation, and intradialytic endothelin-1 (ET-1). Using linear mixed-models, we compared BP slopes during the following intervals: 1-24 hours post-dialysis, 25-44 hours post-dialysis, and 1-44 hours post-dialysis.
Results
There were 25 case subjects with IH and 24 controls. Systolic BP during hours 1-44, 1-24, and 25-44 were 143.1 (16.5), 138.0 (21.2), and 150.8 (22.3) mmHg in controls. For IH subjects, they were 155.4 (14.2), 152.7 (22.8), and 156.5 (20.8) mmHg (p=0.008, 0.02, 0.4). In controls, the slopes were +0.6, +0.6, and +0.4 mmHg/hr. In IH subjects, they were +0.1, -0.3, and +0.3 mmHg/hr. The IH 1-24 hour slope differed from the IH 25-44 hour slope (p=0.001) and the control 1-24 hour slope (p=0.002). The change in ET-1 from pre to post dialysis was 0.5 (1.5) pg/mL in controls and 1.0 (2.3) pg/mL in IH patients (p=0.4). In a univariate model, there was an association with screening BP and BP slope (p=0.002 for controls and p=0.1 for IH patients).
Conclusion
Interdialytic BP patterns differ in IH patients and hemodialysis controls. The elevated post dialysis blood pressure persists for many hours in IH patients contributing to the overall increased BP burden.
Crowdsourced estimation of cognitive decline and resilience in Alzheimer's disease.
Abstract
Identifying accurate biomarkers of cognitive decline is essential for advancing early diagnosis and prevention therapies in Alzheimer's disease. The Alzheimer's disease DREAM Challenge was designed as a computational crowdsourced project to benchmark the current state-of-the-art in predicting cognitive outcomes in Alzheimer's disease based on high dimensional, publicly available genetic and structural imaging data. This meta-analysis failed to identify a meaningful predictor developed from either data modality, suggesting that alternate approaches should be considered for prediction of cognitive performance.
Multisite evaluations of a T2 -relaxation-under-spin-tagging (TRUST) MRI technique to measure brain oxygenation.
Abstract
Purpose
Venous oxygenation (Yv ) is an important index of brain physiology and may be indicative of brain diseases. A T2 -relaxation-under-spin-tagging (TRUST) MRI technique was recently developed to measure Yv . A multisite evaluation of this technique would be an important step toward broader availability and potential clinical utilizations of Yv measures.
Methods
TRUST MRI was performed on a total of 250 healthy subjects, 125 from the developer's site and 25 each from five other sites. All sites were equipped with a 3 Tesla (T) MRI of the same vendor. The estimated Yv and the standard error (SE) of the estimation εYv were compared across sites.
Results
The averaged Yv and εYv across six sites were 61.1% ± 1.4% and 1.3% ± 0.2%, respectively. Multivariate regression analysis showed that the estimated Yv was dependent on age (P = 0.009) but not on performance site. In contrast, the SE of the Yv estimation was site-dependent (P = 0.024) but was less than 1.5%. Further analysis revealed that εYv was positively associated with the amount of subject motion (P < 0.001) but negatively associated with blood signal intensity (P < 0.001).
Conclusion
This work suggests that TRUST MRI can yield equivalent results of Yv estimation across different sites.
KDM4/JMJD2 Histone Demethylase Inhibitors Block Prostate Tumor Growth by Suppressing the Expression of AR and BMYB-Regulated Genes.
Abstract
Histone lysine demethylase KDM4/JMJD2s are overexpressed in many human tumors including prostate cancer (PCa). KDM4s are co-activators of androgen receptor (AR) and are thus potential therapeutic targets. Yet to date few KDM4 inhibitors that have anti-prostate tumor activity in vivo have been developed. Here, we report the anti-tumor growth effect and molecular mechanisms of three novel KDM4 inhibitors (A1, I9, and B3). These inhibitors repressed the transcription of both AR and BMYB-regulated genes. Compound B3 is highly selective for a variety of cancer cell lines including PC3 cells that lack AR. B3 inhibited the in vivo growth of tumors derived from PC3 cells and ex vivo human PCa explants. We identified a novel mechanism by which KDM4B activates the transcription of Polo-like kinase 1 (PLK1). B3 blocked the binding of KDM4B to the PLK1 promoter. Our studies suggest a potential mechanism-based therapeutic strategy for PCa and tumors with elevated KDM4B/PLK1 expression.
Prediction of human population responses to toxic compounds by a collaborative competition.
Abstract
The ability to computationally predict the effects of toxic compounds on humans could help address the deficiencies of current chemical safety testing. Here, we report the results from a community-based DREAM challenge to predict toxicities of environmental compounds with potential adverse health effects for human populations. We measured the cytotoxicity of 156 compounds in 884 lymphoblastoid cell lines for which genotype and transcriptional data are available as part of the Tox21 1000 Genomes Project. The challenge participants developed algorithms to predict interindividual variability of toxic response from genomic profiles and population-level cytotoxicity data from structural attributes of the compounds. 179 submitted predictions were evaluated against an experimental data set to which participants were blinded. Individual cytotoxicity predictions were better than random, with modest correlations (Pearson's r < 0.28), consistent with complex trait genomic prediction. In contrast, predictions of population-level response to different compounds were higher (r < 0.66). The results highlight the possibility of predicting health risks associated with unknown compounds, although risk estimation accuracy remains suboptimal.
Identifying CDKN3 Gene Expression as a Prognostic Biomarker in Lung Adenocarcinoma via Meta-analysis.
Abstract
Lung cancer is among the major causes of cancer deaths, and the survival rate of lung cancer patients is extremely low. Recent studies have demonstrated that the gene CDKN3 is related to neoplasia, but in the literature severe controversy exists over whether it is involved in cancer progression or, conversely, tumor inhibition. In this study, we investigated the expression of CDKN3 and its association with prognosis in lung adenocarcinoma (ADC) and squamous cell carcinoma (SCC) using datasets in Lung Cancer Explorer (LCE; http://qbrc.swmed.edu/lce/). We found that CDKN3 was up-regulated in ADC and SCC compared to normal tissues. We also found that CDKN3 was expressed at a higher level in SCC than in ADC, which was further validated through meta-analysis (coefficient = 2.09, 95% CI = 1.50-2.67, P < 0.0001). In addition, based on meta-analysis for the prognostic value of CDKN3, we found that higher CDKN3 expression was associated with poorer survival outcomes in ADC (HR = 1.65, 95% CI = 1.39-1.96, P < 0.0001), but not in SCC (HR = 1.10, 95% CI = 0.84-1.44, P = 0.494). Our findings indicate that CDKN3 may be a prognostic marker in ADC, though the detailed mechanism is yet to be revealed.
Nonoptimal codon usage influences protein structure in intrinsically disordered regions.
Abstract
Synonymous codons are not used with equal frequencies in most genomes. Codon usage has been proposed to play a role in regulating translation kinetics and co-translational protein folding. The relationship between codon usage and protein structures and the in vivo role of codon usage in eukaryotic protein folding is not clear. Here, we show that there is a strong codon usage bias in the filamentous fungus Neurospora. Importantly, we found genome-wide correlations between codon choices and predicted protein secondary structures: Nonoptimal codons are preferentially used in intrinsically disordered regions, and more optimal codons are used in structured domains. The functional importance of such correlations in vivo was confirmed by structure-based codon manipulation of codons in the Neurospora circadian clock gene frequency (frq). The codon optimization of the predicted disordered, but not well-structured regions of FRQ impairs clock function and altered FRQ structures. Furthermore, the correlations between codon usage and protein disorder tendency are conserved in other eukaryotes. Together, these results suggest that codon choices and protein structures co-evolve to ensure proper protein folding in eukaryotic organisms.
Design and bioinformatics analysis of genome-wide CLIP experiments.
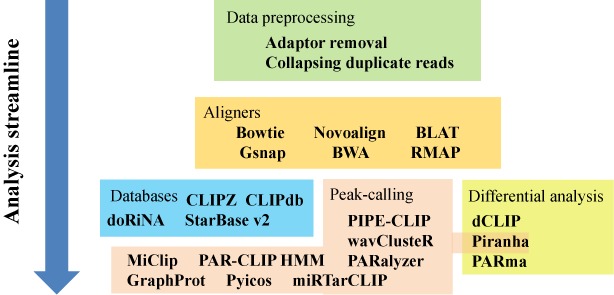
Abstract
The past decades have witnessed a surge of discoveries revealing RNA regulation as a central player in cellular processes. RNAs are regulated by RNA-binding proteins (RBPs) at all post-transcriptional stages, including splicing, transportation, stabilization and translation. Defects in the functions of these RBPs underlie a broad spectrum of human pathologies. Systematic identification of RBP functional targets is among the key biomedical research questions and provides a new direction for drug discovery. The advent of cross-linking immunoprecipitation coupled with high-throughput sequencing (genome-wide CLIP) technology has recently enabled the investigation of genome-wide RBP-RNA binding at single base-pair resolution. This technology has evolved through the development of three distinct versions: HITS-CLIP, PAR-CLIP and iCLIP. Meanwhile, numerous bioinformatics pipelines for handling the genome-wide CLIP data have also been developed. In this review, we discuss the genome-wide CLIP technology and focus on bioinformatics analysis. Specifically, we compare the strengths and weaknesses, as well as the scopes, of various bioinformatics tools. To assist readers in choosing optimal procedures for their analysis, we also review experimental design and procedures that affect bioinformatics analyses.
Statistical completion of a partially identified graph with applications for the estimation of gene regulatory networks.
Abstract
We study the estimation of a Gaussian graphical model whose dependent structures are partially identified. In a Gaussian graphical model, an off-diagonal zero entry in the concentration matrix (the inverse covariance matrix) implies the conditional independence of two corresponding variables, given all other variables. A number of methods have been proposed to estimate a sparse large-scale Gaussian graphical model or, equivalently, a sparse large-scale concentration matrix. In practice, the graph structure to be estimated is often partially identified by other sources or a pre-screening. In this paper, we propose a simple modification of existing methods to take into account this information in the estimation. We show that the partially identified dependent structure reduces the error in estimating the dependent structure. We apply the proposed method to estimating the gene regulatory network from lung cancer data, where protein-protein interactions are partially identified from the human protein reference database. The application shows that proposed method identified many important cancer genes as hub genes in the constructed lung cancer network. In addition, we validated the prognostic importance of a newly identified cancer gene, PTPN13, in four independent lung cancer datasets. The results indicate that the proposed method could facilitate studying underlying lung cancer mechanisms and identifying reliable biomarkers for lung cancer prognosis.
Decreased BECN1 mRNA Expression in Human Breast Cancer is Associated with Estrogen Receptor-Negative Subtypes and Poor Prognosis.
Abstract
Both BRCA1 and Beclin 1 (BECN1) are tumor suppressor genes, which are in close proximity on the human chromosome 17q21 breast cancer tumor susceptibility locus and are often concurrently deleted. However, their importance in sporadic human breast cancer is not known. To interrogate the effects of BECN1 and BRCA1 in breast cancer, we studied their mRNA expression patterns in breast cancer patients from two large datasets: The Cancer Genome Atlas (TCGA) (n=1067) and the Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) (n=1992). In both datasets, low expression of BECN1 was more common in HER2-enriched and basal-like (mostly triple-negative) breast cancers compared to luminal A/B intrinsic tumor subtypes, and was also strongly associated with TP53 mutations and advanced tumor grade. In contrast, there was no significant association between low BRCA1 expression and HER2-enriched or basal-like subtypes, TP53 mutations or tumor grade. In addition, low expression of BECN1 (but not low BRCA1) was associated with poor prognosis, and BECN1 (but not BRCA1) expression was an independent predictor of survival. These findings suggest that decreased mRNA expression of the autophagy gene BECN1 may contribute to the pathogenesis and progression of HER2-enriched, basal-like, and TP53 mutant breast cancers.
iScreen: Image-Based High-Content RNAi Screening Analysis Tools.
Abstract
High-throughput RNA interference (RNAi) screening has opened up a path to investigating functional genomics in a genome-wide pattern. However, such studies are often restricted to assays that have a single readout format. Recently, advanced image technologies have been coupled with high-throughput RNAi screening to develop high-content screening, in which one or more cell image(s), instead of a single readout, were generated from each well. This image-based high-content screening technology has led to genome-wide functional annotation in a wider spectrum of biological research studies, as well as in drug and target discovery, so that complex cellular phenotypes can be measured in a multiparametric format. Despite these advances, data analysis and visualization tools are still largely lacking for these types of experiments. Therefore, we developed iScreen (image-Based High-content RNAi Screening Analysis Tool), an R package for the statistical modeling and visualization of image-based high-content RNAi screening. Two case studies were used to demonstrate the capability and efficiency of the iScreen package. iScreen is available for download on CRAN (http://cran.cnr.berkeley.edu/web/packages/iScreen/index.html). The user manual is also available as a supplementary document.
Acute effect of glucose on cerebral blood flow, blood oxygenation, and oxidative metabolism.
Abstract
While it is known that specific nuclei of the brain, for example hypothalamus, contain glucose-sensing neurons thus their activity is affected by blood glucose level, the effect of glucose modulation on whole-brain metabolism is not completely understood. Several recent reports have elucidated the long-term impact of caloric restriction on the brain, showing that animals under caloric restriction had enhanced rate of tricarboxylic acid cycle (TCA) cycle flux accompanied by extended life span. However, acute effect of postprandial blood glucose increase has not been addressed in detail, partly due to a scarcity and complexity of measurement techniques. In this study, using a recently developed noninvasive MR technique, we measured dynamic changes in global cerebral metabolic rate of O2 (CMRO2 ) following a 50 g glucose ingestion (N = 10). A time dependent decrease in CMRO2 was observed, which was accompanied by a reduction in oxygen extraction fraction (OEF) with unaltered cerebral blood flow (CBF). At 40 min post-ingestion, the amount of CMRO2 reduction was 7.8 ± 1.6%. A control study without glucose ingestion was performed (N = 10), which revealed no changes in CMRO2 , CBF, or OEF, suggesting that the observations in the glucose study was not due to subject drowsiness or fatigue after staying inside the scanner. These findings suggest that ingestion of glucose may alter the rate of cerebral metabolism of oxygen in an acute setting.
A community computational challenge to predict the activity of pairs of compounds.
Abstract
Recent therapeutic successes have renewed interest in drug combinations, but experimental screening approaches are costly and often identify only small numbers of synergistic combinations. The DREAM consortium launched an open challenge to foster the development of in silico methods to computationally rank 91 compound pairs, from the most synergistic to the most antagonistic, based on gene-expression profiles of human B cells treated with individual compounds at multiple time points and concentrations. Using scoring metrics based on experimental dose-response curves, we assessed 32 methods (31 community-generated approaches and SynGen), four of which performed significantly better than random guessing. We highlight similarities between the methods. Although the accuracy of predictions was not optimal, we find that computational prediction of compound-pair activity is possible, and that community challenges can be useful to advance the field of in silico compound-synergy prediction.
Ensemble-based network aggregation improves the accuracy of gene network reconstruction.
Abstract
Reverse engineering approaches to constructing gene regulatory networks (GRNs) based on genome-wide mRNA expression data have led to significant biological findings, such as the discovery of novel drug targets. However, the reliability of the reconstructed GRNs needs to be improved. Here, we propose an ensemble-based network aggregation approach to improving the accuracy of network topologies constructed from mRNA expression data. To evaluate the performances of different approaches, we created dozens of simulated networks from combinations of gene-set sizes and sample sizes and also tested our methods on three Escherichia coli datasets. We demonstrate that the ensemble-based network aggregation approach can be used to effectively integrate GRNs constructed from different studies - producing more accurate networks. We also apply this approach to building a network from epithelial mesenchymal transition (EMT) signature microarray data and identify hub genes that might be potential drug targets. The R code used to perform all of the analyses is available in an R package entitled "ENA", accessible on CRAN (http://cran.r-project.org/web/packages/ENA/).
Transcriptional interference by antisense RNA is required for circadian clock function.
Abstract
Eukaryotic circadian oscillators consist of negative feedback loops that generate endogenous rhythmicities. Natural antisense RNAs are found in a wide range of eukaryotic organisms. Nevertheless, the physiological importance and mode of action of most antisense RNAs are not clear. frequency (frq) encodes a component of the Neurospora core circadian negative feedback loop, which was thought to generate sustained rhythmicity. Transcription of qrf, the long non-coding frq antisense RNA, is induced by light, and its level oscillates in antiphase to frq sense RNA. Here we show that qrf transcription is regulated by both light-dependent and light-independent mechanisms. Light-dependent qrf transcription represses frq expression and regulates clock resetting. Light-independent qrf expression, on the other hand, is required for circadian rhythmicity. frq transcription also inhibits qrf expression and drives the antiphasic rhythm of qrf transcripts. The mutual inhibition of frq and qrf transcription thus forms a double negative feedback loop that is interlocked with the core feedback loop. Genetic and mathematical modelling analyses indicate that such an arrangement is required for robust and sustained circadian rhythmicity. Moreover, our results suggest that antisense transcription inhibits sense expression by mediating chromatin modifications and premature termination of transcription. Taken together, our results establish antisense transcription as an essential feature in a circadian system and shed light on the importance and mechanism of antisense action.
Predictors and intensity of online access to electronic medical records among patients with cancer.
Abstract
Introduction
Electronic portals are secure Web-based servers that provide patients with real-time access to their personal health record (PHR). These applications are now widely used at cancer centers nationwide, but their impact has not been well studied. This study set out to determine predictors and patterns of use of a Web-based portal for accessing PHRs and communicating with health providers among patients with cancer.
Methods
Retrospective analysis of enrollment in and use of MyChart, a PHR portal for the Epic electronic medical record system, among patients seen at a National Cancer Institute-designated cancer center. Predictors of MyChart use were analyzed through univariable and multivariable regression models.
Results
A total of 6,495 patients enrolled in MyChart from 2007 to 2012. The median number of log-ins over this period was 57 (interquartile range 17-137). The most common portal actions were viewing test results (37%), viewing and responding to clinic messages (29%), and sending medical advice requests (6.4%). Increased portal use was significantly associated with younger age, white race, and an upper aerodigestive malignancy diagnosis. Thirty-seven percent of all log-ins and 31% of all medical advice requests occurred outside clinic hours. Over the study period, the average number of patient log-ins per year more than doubled.
Conclusions
Among patients with cancer, PHR portal use is frequent and increasing. Younger patients, white patients, and patients with upper aerodigestive malignancies exhibit the heaviest portal use. Understanding the implications of this new technology will be central to the delivery of safe and effective care.
Autophagy is required for G₁/G₀ quiescence in response to nitrogen starvation in Saccharomyces cerevisiae.
Abstract
In response to starvation, cells undergo increased levels of autophagy and cell cycle arrest but the role of autophagy in starvation-induced cell cycle arrest is not fully understood. Here we show that autophagy genes regulate cell cycle arrest in the budding yeast Saccharomyces cerevisiae during nitrogen starvation. While exponentially growing wild-type yeasts preferentially arrest in G₁/G₀ in response to starvation, yeasts carrying null mutations in autophagy genes show a significantly higher percentage of cells in G₂/M. In these autophagy-deficient yeast strains, starvation elicits physiological properties associated with quiescence, such as Snf1 activation, glycogen and trehalose accumulation as well as heat-shock resistance. However, while nutrient-starved wild-type yeasts finish the G₂/M transition and arrest in G₁/G 0₀ autophagy-deficient yeasts arrest in telophase. Our results suggest that autophagy is crucial for mitotic exit during starvation and appropriate entry into a G₁/G₀ quiescent state.
The role of homeostatic regulation between tumor suppressor DAB2IP and oncogenic Skp2 in prostate cancer growth.
Abstract
Altered DAB2IP gene expression often detected in prostate cancer (PCa) is due to epigenetic silencing. In this study, we unveil a new mechanism leading to the loss of DAB2IP protein; an oncogenic S-phase kinase-associated protein-2 (Skp2) as E3 ubiquitin ligase plays a key regulator in DAB2IP degradation. In order to unveil the role of Skp2 in the turnover of DAB2IP protein, both prostate cell lines and prostate cancer specimens with a variety of molecular and cell biologic techniques were employed. We demonstrated that DAB2IP is regulated by Skp2-mediated proteasome degradation in the prostate cell lines. Further analyses identified the N-terminal DAB2IP containing the ubiquitination site. Immunohistochemical study exhibited an inverse correlation between DAB2IP and Skp2 protein expression in the prostate cancer tissue microarray. In contrast, DAB2IP can suppressSkp2 protein expression is mediated through Akt signaling. The reciprocal regulation between DAB2IP and Skp2 can impact on the growth of PCa cells. This reciprocal regulation between DAB2IP and Skp2 protein represents a unique homeostatic balance between tumor suppressor and oncoprotein in normal prostate epithelia, which is apparently altered in cancer cells. The outcome of this study has identified new potential targets for developing new therapeutic strategy for PCa.
Molecular markers of carcinogenesis for risk stratification of individuals with colorectal polyps: a case-control study.
Abstract
Risk stratification using number, size, and histology of colorectal adenomas is currently suboptimal for identifying patients at increased risk for future colorectal cancer. We hypothesized that molecular markers of carcinogenesis in adenomas, measured via immunohistochemistry, may help identify high-risk patients. To test this hypothesis, we conducted a retrospective, 1:1 matched case-control study (n = 216; 46% female) in which cases were patients with colorectal cancer and synchronous adenoma and controls were patients with adenoma but no colorectal cancer at baseline or within 5 years of follow-up. In phase I of analyses, we compared expression of molecular markers of carcinogenesis in case and control adenomas, blind to case status. In phase II of analyses, patients were randomly divided into independent training and validation groups to develop a model for predicting case status. We found that seven markers [p53, p21, Cox-2, β-catenin (BCAT), DNA-dependent protein kinase (DNApkcs), survivin, and O6-methylguanine-DNA methyltransferase (MGMT)] were significantly associated with case status on unadjusted analyses, as well as analyses adjusted for age and advanced adenoma status (P < 0.01 for at least one marker component). When applied to the validation set, a predictive model using these seven markers showed substantial accuracy for identifying cases [area under the receiver operation characteristic curve (AUC), 0.83; 95% confidence interval (CI), 0.74-0.92]. A parsimonious model using three markers performed similarly to the seven-marker model (AUC, 0.84). In summary, we found that molecular markers of carcinogenesis distinguished adenomas from patients with and without colorectal cancer. Furthermore, we speculate that prospective studies using molecular markers to identify individuals with polyps at risk for future neoplasia are warranted.
Validation of a serum screen for Alzheimer's disease across assay platforms, species, and tissues.
Abstract
Background
There is a significant need for rapid and cost-effective biomarkers of Alzheimer's disease (AD) for advancement of clinical practice and therapeutic trials.
Objective
The aim of the current study was to cross-validate our previously published serum-based algorithm on an independent assay platform as well as validate across tissues and species. Preliminary analyses were conducted to examine the utility in distinguishing AD from non-AD neurological disease (Parkinson's disease, PD).
Methods
Serum proteins from our previously published algorithm were quantified from 150 AD cases and 150 controls on the Meso Scale Discovery (MSD) platform. Serum samples were analyzed from 49 PD cases and compared to a random sample of 51 AD cases and 62 controls. Support vector machines (SVM) were used to discriminate PD versus AD versus controls. Human and AD mouse model microvessel images were quantified with HAMAMATSU imaging software. Mouse serum biomarkers were assayed via MSD.
Results
Analysis of 21 serum proteins from 150 AD cases and 150 controls yielded an algorithm with sensitivity and specificity of 0.90 for correctly classifying AD. This multi-marker approach was then validated across species and tissue. Assay of the top proteins in human and AD mouse model brain microvessels correctly classified 90-100% of the samples. SVM analyses were highly accurate at distinguishing PD versus AD versus controls.
Conclusions
This serum-based biomarker panel should be tested in a community-based setting to determine its utility as a first-line screen for AD and non-AD neurological diseases for primary care providers.
Computational detection and suppression of sequence-specific off-target phenotypes from whole genome RNAi screens.
Abstract
challenge for large-scale siRNA loss-of-function studies is the biological pleiotropy resulting from multiple modes of action of siRNA reagents. A major confounding feature of these reagents is the microRNA-like translational quelling resulting from short regions of oligonucleotide complementarity to many different messenger RNAs. We developed a computational approach, deconvolution analysis of RNAi screening data, for automated quantitation of off-target effects in RNAi screening data sets. Substantial reduction of off-target rates was experimentally validated in five distinct biological screens across different genome-wide siRNA libraries. A public-access graphical-user-interface has been constructed to facilitate application of this algorithm.
A model-based approach to identify binding sites in CLIP-Seq data.
Abstract
Cross-linking immunoprecipitation coupled with high-throughput sequencing (CLIP-Seq) has made it possible to identify the targeting sites of RNA-binding proteins in various cell culture systems and tissue types on a genome-wide scale. Here we present a novel model-based approach (MiClip) to identify high-confidence protein-RNA binding sites from CLIP-seq datasets. This approach assigns a probability score for each potential binding site to help prioritize subsequent validation experiments. The MiClip algorithm has been tested in both HITS-CLIP and PAR-CLIP datasets. In the HITS-CLIP dataset, the signal/noise ratios of miRNA seed motif enrichment produced by the MiClip approach are between 17% and 301% higher than those by the ad hoc method for the top 10 most enriched miRNAs. In the PAR-CLIP dataset, the MiClip approach can identify ∼50% more validated binding targets than the original ad hoc method and two recently published methods. To facilitate the application of the algorithm, we have released an R package, MiClip (http://cran.r-project.org/web/packages/MiClip/index.html), and a public web-based graphical user interface software (http://galaxy.qbrc.org/tool_runner?tool_id=mi_clip) for customized analysis.
Bayesian hidden Markov models to identify RNA-protein interaction sites in PAR-CLIP.
Abstract
The photoactivatable ribonucleoside enhanced cross-linking immunoprecipitation (PAR-CLIP) has been increasingly used for the global mapping of RNA-protein interaction sites. There are two key features of the PAR-CLIP experiments: The sequence read tags are likely to form an enriched peak around each RNA-protein interaction site; and the cross-linking procedure is likely to introduce a specific mutation in each sequence read tag at the interaction site. Several ad hoc methods have been developed to identify the RNA-protein interaction sites using either sequence read counts or mutation counts alone; however, rigorous statistical methods for analyzing PAR-CLIP are still lacking. In this article, we propose an integrative model to establish a joint distribution of observed read and mutation counts. To pinpoint the interaction sites at single base-pair resolution, we developed a novel modeling approach that adopts non-homogeneous hidden Markov models to incorporate the nucleotide sequence at each genomic location. Both simulation studies and data application showed that our method outperforms the ad hoc methods, and provides reliable inferences for the RNA-protein binding sites from PAR-CLIP data.
dCLIP: a computational approach for comparative CLIP-seq analyses.
Abstract
Although comparison of RNA-protein interaction profiles across different conditions has become increasingly important to understanding the function of RNA-binding proteins (RBPs), few computational approaches have been developed for quantitative comparison of CLIP-seq datasets. Here, we present an easy-to-use command line tool, dCLIP, for quantitative CLIP-seq comparative analysis. The two-stage method implemented in dCLIP, including a modified MA normalization method and a hidden Markov model, is shown to be able to effectively identify differential binding regions of RBPs in four CLIP-seq datasets, generated by HITS-CLIP, iCLIP and PAR-CLIP protocols. dCLIP is freely available at http://qbrc.swmed.edu/software/.
Detection of candidate tumor driver genes using a fully integrated Bayesian approach.
Abstract
DNA copy number alterations (CNAs), including amplifications and deletions, can result in significant changes in gene expression and are closely related to the development and progression of many diseases, especially cancer. For example, CNA-associated expression changes in certain genes (called candidate tumor driver genes) can alter the expression levels of many downstream genes through transcription regulation and cause cancer. Identification of such candidate tumor driver genes leads to discovery of novel therapeutic targets for personalized treatment of cancers. Several approaches have been developed for this purpose by using both copy number and gene expression data. In this study, we propose a Bayesian approach to identify candidate tumor driver genes, in which the copy number and gene expression data are modeled together, and the dependency between the two data types is modeled through conditional probabilities. The proposed joint modeling approach can identify CNA and differentially expressed genes simultaneously, leading to improved detection of candidate tumor driver genes and comprehensive understanding of underlying biological processes. We evaluated the proposed method in simulation studies, and then applied to a head and neck squamous cell carcinoma data set. Both simulation studies and data application show that the joint modeling approach can significantly improve the performance in identifying candidate tumor driver genes, when compared with other existing approaches.
Adaptive prediction model in prospective molecular signature-based clinical studies.
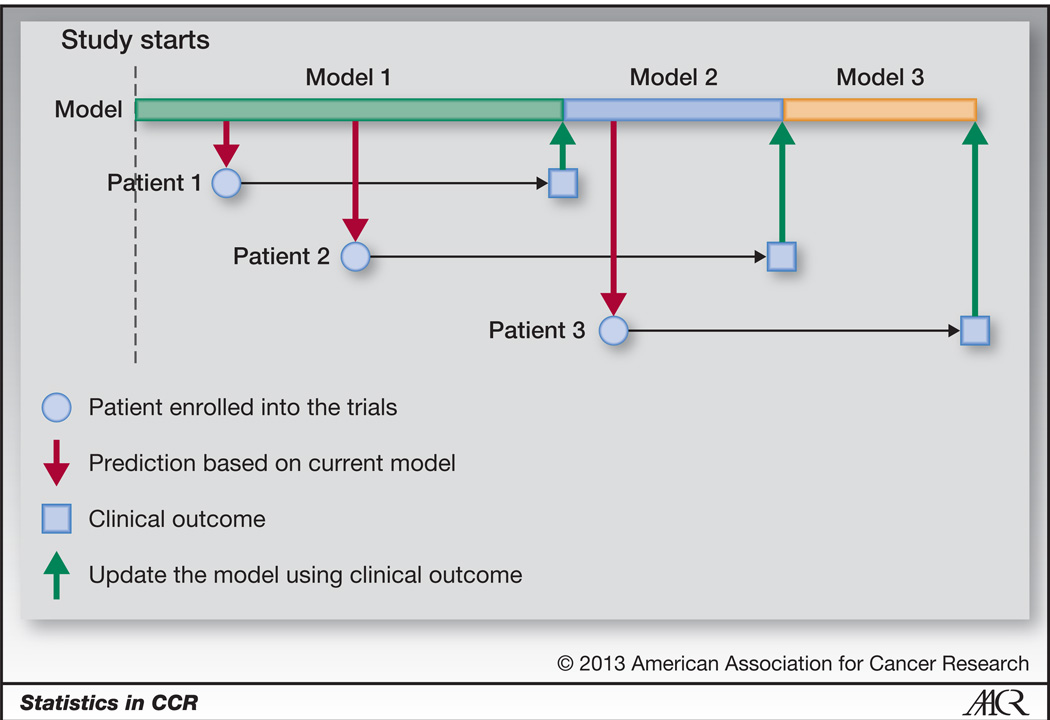
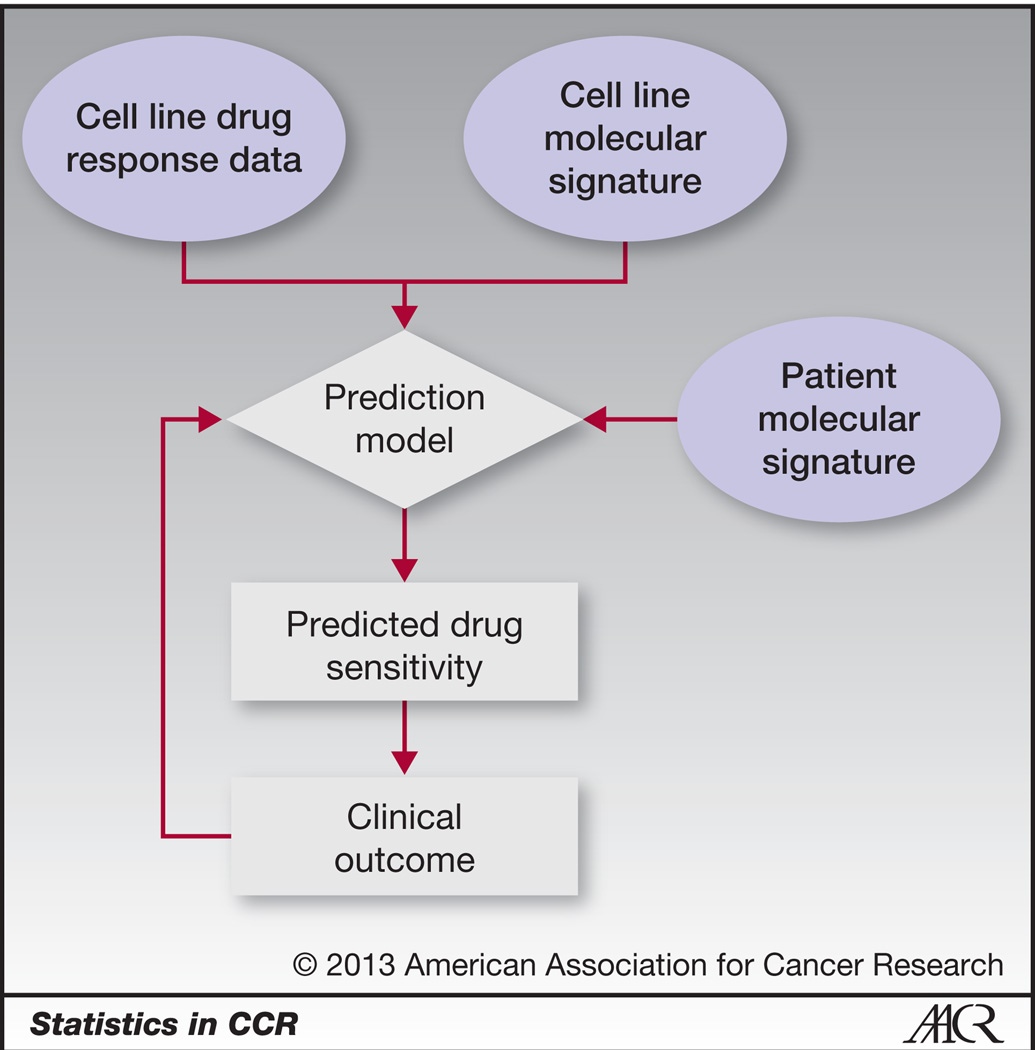
Abstract
Use of molecular profiles and clinical information can help predict which treatment would give the best outcome and survival for each individual patient, and thus guide optimal therapy, which offers great promise for the future of clinical trials and practice. High prediction accuracy is essential for selecting the best treatment plan. The gold standard for evaluating the prediction models is prospective clinical studies, in which patients are enrolled sequentially. However, there is no statistical method using this sequential feature to adapt the prediction model to the current patient cohort. In this article, we propose a reweighted random forest (RWRF) model, which updates the weight of each decision tree whenever additional patient information is available, to account for the potential heterogeneity between training and testing data. A simulation study and a lung cancer example are used to show that the proposed method can adapt the prediction model to current patients' characteristics, and, therefore, can improve prediction accuracy significantly. We also show that the proposed method can identify important and consistent predictive variables. Compared with rebuilding the prediction model, the RWRF updates a well-tested model gradually, and all of the adaptive procedure/parameters used in the RWRF model are prespecified before patient recruitment, which are important practical advantages for prospective clinical studies.
Molecular neuropsychology: creation of test-specific blood biomarker algorithms.
Abstract
Background
Prior work on the link between blood-based biomarkers and cognitive status has largely been based on dichotomous classifications rather than detailed neuropsychological functioning. The current project was designed to create serum-based biomarker algorithms that predict neuropsychological test performance.
Methods
A battery of neuropsychological measures was administered. Random forest analyses were utilized to create neuropsychological test-specific biomarker risk scores in a training set that were entered into linear regression models predicting the respective test scores in the test set. Serum multiplex biomarker data were analyzed on 108 proteins from 395 participants (197 Alzheimer patients and 198 controls) from the Texas Alzheimer's Research and Care Consortium.
Results
The biomarker risk scores were significant predictors (p < 0.05) of scores on all neuropsychological tests. With the exception of premorbid intellectual status (6.6%), the biomarker risk scores alone accounted for a minimum of 12.9% of the variance in neuropsychological scores. Biomarker algorithms (biomarker risk scores and demographics) accounted for substantially more variance in scores. Review of the variable importance plots indicated differential patterns of biomarker significance for each test, suggesting the possibility of domain-specific biomarker algorithms.
Conclusions
Our findings provide proof of concept for a novel area of scientific discovery, which we term 'molecular neuropsychology'.
Review of biological network data and its applications.
Abstract
Studying biological networks, such as protein-protein interactions, is key to understanding complex biological activities. Various types of large-scale biological datasets have been collected and analyzed with high-throughput technologies, including DNA microarray, next-generation sequencing, and the two-hybrid screening system, for this purpose. In this review, we focus on network-based approaches that help in understanding biological systems and identifying biological functions. Accordingly, this paper covers two major topics in network biology: reconstruction of gene regulatory networks and network-based applications, including protein function prediction, disease gene prioritization, and network-based genome-wide association study.
Using functional signature ontology (FUSION) to identify mechanisms of action for natural products.
Abstract
A challenge for biomedical research is the development of pharmaceuticals that appropriately target disease mechanisms. Natural products can be a rich source of bioactive chemicals for medicinal applications but can act through unknown mechanisms and can be difficult to produce or obtain. To address these challenges, we developed a new marine-derived, renewable natural products resource and a method for linking bioactive derivatives of this library to the proteins and biological processes that they target in cells. We used cell-based screening and computational analysis to match gene expression signatures produced by natural products to those produced by small interfering RNA (siRNA) and synthetic microRNA (miRNA) libraries. With this strategy, we matched proteins and miRNAs with diverse biological processes and also identified putative protein targets and mechanisms of action for several previously undescribed marine-derived natural products. We confirmed mechanistic relationships for selected siRNAs, miRNAs, and compounds with functional roles in autophagy, chemotaxis mediated by discoidin domain receptor 2, or activation of the kinase AKT. Thus, this approach may be an effective method for screening new drugs while simultaneously identifying their targets.
EGFR-mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance.
Abstract
Cell surface growth factor receptors couple environmental cues to the regulation of cytoplasmic homeostatic processes, including autophagy, and aberrant activation of such receptors is a common feature of human malignancies. Here, we defined the molecular basis by which the epidermal growth factor receptor (EGFR) tyrosine kinase regulates autophagy. Active EGFR binds the autophagy protein Beclin 1, leading to its multisite tyrosine phosphorylation, enhanced binding to inhibitors, and decreased Beclin 1-associated VPS34 kinase activity. EGFR tyrosine kinase inhibitor (TKI) therapy disrupts Beclin 1 tyrosine phosphorylation and binding to its inhibitors and restores autophagy in non-small-cell lung carcinoma (NSCLC) cells with a TKI-sensitive EGFR mutation. In NSCLC tumor xenografts, the expression of a tyrosine phosphomimetic Beclin 1 mutant leads to reduced autophagy, enhanced tumor growth, tumor dedifferentiation, and resistance to TKI therapy. Thus, oncogenic receptor tyrosine kinases directly regulate the core autophagy machinery, which may contribute to tumor progression and chemoresistance.
Beclin 2 functions in autophagy, degradation of G protein-coupled receptors, and metabolism.
Abstract
The molecular mechanism of autophagy and its relationship to other lysosomal degradation pathways remain incompletely understood. Here, we identified a previously uncharacterized mammalian-specific protein, Beclin 2, which, like Beclin 1, functions in autophagy and interacts with class III PI3K complex components and Bcl-2. However, Beclin 2, but not Beclin 1, functions in an additional lysosomal degradation pathway. Beclin 2 is required for ligand-induced endolysosomal degradation of several G protein-coupled receptors (GPCRs) through its interaction with GASP1. Beclin 2 homozygous knockout mice have decreased embryonic viability, and heterozygous knockout mice have defective autophagy, increased levels of brain cannabinoid 1 receptor, elevated food intake, and obesity and insulin resistance. Our findings identify Beclin 2 as a converging regulator of autophagy and GPCR turnover and highlight the functional and mechanistic diversity of Beclin family members in autophagy, endolysosomal trafficking, and metabolism.
SbacHTS: spatial background noise correction for high-throughput RNAi screening.
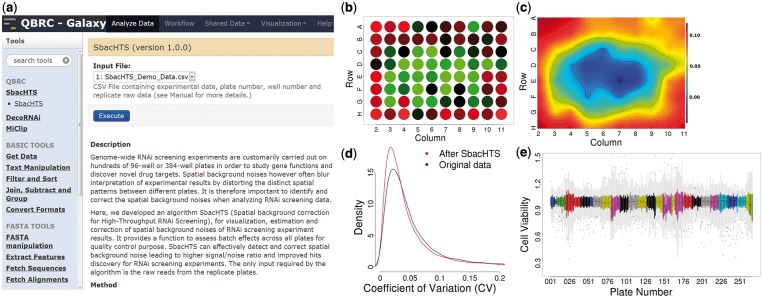
Abstract
Motivation
High-throughput cell-based phenotypic screening has become an increasingly important technology for discovering new drug targets and assigning gene functions. Such experiments use hundreds of 96-well or 384-well plates, to cover whole-genome RNAi collections and/or chemical compound files, and often collect measurements that are sensitive to spatial background noise whose patterns can vary across individual plates. Correcting these position effects can substantially improve measurement accuracy and screening success.
Result
We developed SbacHTS (Spatial background noise correction for High-Throughput RNAi Screening) software for visualization, estimation and correction of spatial background noise in high-throughput RNAi screens. SbacHTS is supported on the Galaxy open-source framework with a user-friendly open access web interface. We find that SbacHTS software can effectively detect and correct spatial background noise, increase signal to noise ratio and enhance statistical detection power in high-throughput RNAi screening experiments.
Availability
http://www.galaxy.qbrc.org/Alterations in resting functional connectivity due to recent motor task.
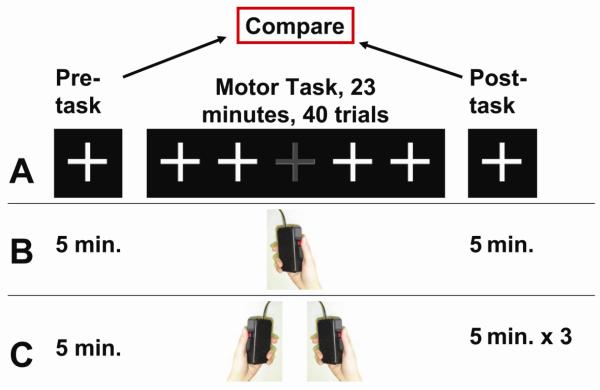
Abstract
The impact of recent experiences of task performance on resting functional connectivity MRI (fcMRI) has important implications for the design of many neuroimaging studies, because, if an effect is present, the fcMRI scan then must be performed before any evoked fMRI or after a time gap to allow it to dissipate. The present study aims to determine the effect of simple button presses, which are used in many cognitive fMRI tasks as a response recording method, on later acquired fcMRI data. Human volunteers were subject to a 23-minute button press motor task. Their resting-state brain activity before and after the task was assessed with fcMRI. It was found that, compared to the pre-task resting period, the post-task resting fcMRI revealed a significantly higher (p=0.002, N=24) cross correlation coefficient (CC) between left and right motor cortices. These changes were not present in sham control studies that matched the paradigm timing but had no actual task. The amplitude of fcMRI signal fluctuation (AF) also demonstrated an increase in the post-task period compared to pre-task. These changes were observed using both the right-hand-only task and the two-hand task. Study of the recovery time course of these effects revealed that the CC changes lasted for about 5 min while the AF change lasted for at least 15 min. Finally, voxelwise analysis revealed that the pre/post-task differences were also observed in several other brain regions, including the auditory cortex, visual areas, and the thalamus. Our data suggest that the recent performance of the simple button press task can result in elevated fcMRI CC and AF in relevant brain networks and that fcMRI scan should be performed either before evoked fMRI or after a sufficient time gap following fMRI.
A powerful Bayesian meta-analysis method to integrate multiple gene set enrichment studies.
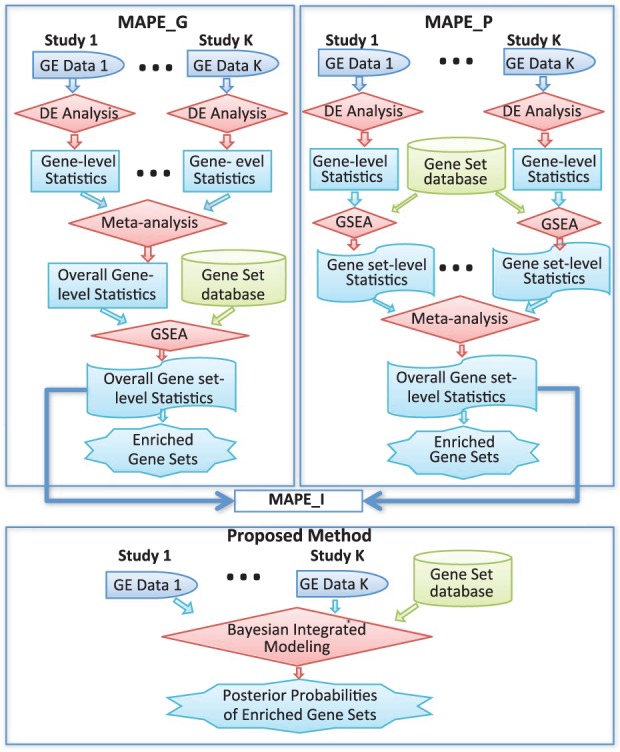
Abstract
Motivation
Much research effort has been devoted to the identification of enriched gene sets for microarray experiments. However, identified gene sets are often found to be inconsistent among independent studies. This is probably owing to the noisy data of microarray experiments coupled with small sample sizes of individual studies. Therefore, combining information from multiple studies is likely to improve the detection of truly enriched gene classes. As more and more data become available, it calls for statistical methods to integrate information from multiple studies, also known as meta-analysis, to improve the power of identifying enriched gene sets.
Results
Results
We propose a Bayesian model that provides a coherent framework for joint modeling of both gene set information and gene expression data from multiple studies, to improve the detection of enriched gene sets by leveraging information from different sources available. One distinct feature of our method is that it directly models the gene expression data, instead of using summary statistics, when synthesizing studies. Besides, the proposed model is flexible and offers an appropriate treatment of between-study heterogeneities that frequently arise in the meta-analysis of microarray experiments. We show that under our Bayesian model, the full posterior conditionals all have known distributions, which greatly facilitates the MCMC computation. Simulation results show that the proposed method can improve the power of gene set enrichment meta-analysis, as opposed to existing methods developed by Shen and Tseng (2010, Bioinformatics, 26, 1316-1323), and it is not sensitive to mild or moderate deviations from the distributional assumption for gene expression data. We illustrate the proposed method through an application of combining eight lung cancer datasets for gene set enrichment analysis, which demonstrates the usefulness of the method.
Biomarkers of Alzheimer's disease among Mexican Americans.
Abstract
Background
Mexican Americans are the fastest aging segment of the U.S. population, yet little scientific literature exists regarding the Alzheimer's disease (AD) among this segment of the population. The extant literature suggests that biomarkers of AD will vary according to race/ethnicity though no prior work has explicitly studied this possibility. The aim of this study was to create a serum-based biomarker profile of AD among Mexican American.
Methods
Data were analyzed from 363 Mexican American participants (49 AD and 314 normal controls) enrolled in the Texas Alzheimer's Research & Care Consortium (TARCC). Non-fasting serum samples were analyzed using a luminex-based multi-plex platform. A biomarker profile was generated using random forest analyses.
Results
The biomarker profile of AD among Mexican Americans was different from prior work from non-Hispanic populations with regards to the variable importance plots. In fact, many of the top markers were related to metabolic factors (e.g., FABP, GLP-1, CD40, pancreatic polypeptide, insulin-like-growth factor, and insulin). The biomarker profile was a significant classifier of AD status yielding an area under the receiver operating characteristic curve, sensitivity, and specificity of 0.77, 0.92, and 0.64, respectively. Combining biomarkers with clinical variables yielded a better balance of sensitivity and specificity.
Conclusion
The biomarker profile for AD among Mexican American cases is significantly different from that previously identified among non-Hispanic cases from many large-scale studies. This is the first study to explicitly examine and provide support for blood-based biomarkers of AD among Mexican Americans. Areas for future research are highlighted.
A 12-gene set predicts survival benefits from adjuvant chemotherapy in non-small cell lung cancer patients.
Abstract
Purpose
Prospectively identifying who will benefit from adjuvant chemotherapy (ACT) would improve clinical decisions for non-small cell lung cancer (NSCLC) patients. In this study, we aim to develop and validate a functional gene set that predicts the clinical benefits of ACT in NSCLC.
Experimental Design
An 18-hub-gene prognosis signature was developed through a systems biology approach, and its prognostic value was evaluated in six independent cohorts. The 18-hub-gene set was then integrated with genome-wide functional (RNAi) data and genetic aberration data to derive a 12-gene predictive signature for ACT benefits in NSCLC.
Results
Using a cohort of 442 stage I to III NSCLC patients who underwent surgical resection, we identified an 18-hub-gene set that robustly predicted the prognosis of patients with adenocarcinoma in all validation datasets across four microarray platforms. The hub genes, identified through a purely data-driven approach, have significant biological implications in tumor pathogenesis, including NKX2-1, Aurora Kinase A, PRC1, CDKN3, MBIP, and RRM2. The 12-gene predictive signature was successfully validated in two independent datasets (n = 90 and 176). The predicted benefit group showed significant improvement in survival after ACT (UT Lung SPORE data: HR = 0.34, P = 0.017; JBR.10 clinical trial data: HR = 0.36, P = 0.038), whereas the predicted nonbenefit group showed no survival benefit for 2 datasets (HR = 0.80, P = 0.70; HR = 0.91, P = 0.82).
Conclusion
This is the first study to integrate genetic aberration, genome-wide RNAi data, and mRNA expression data to identify a functional gene set that predicts which resectable patients with non-small cell lung cancer will have a survival benefit with ACT.
Epigenetic change detection and pattern recognition via Bayesian hierarchical hidden Markov models.
Abstract
Introduction
Epigenetics is the study of changes to the genome that can switch genes on or off and determine which proteins are transcribed without altering the DNA sequence. Recently, epigenetic changes have been linked to the development and progression of disease such as psychiatric disorders. High-throughput epigenetic experiments have enabled researchers to measure genome-wide epigenetic profiles and yield data consisting of intensity ratios of immunoprecipitation versus reference samples. The intensity ratios can provide a view of genomic regions where protein binding occur under one experimental condition and further allow us to detect epigenetic alterations through comparison between two different conditions. However, such experiments can be expensive, with only a few replicates available. Moreover, epigenetic data are often spatially correlated with high noise levels. In this paper, we develop a Bayesian hierarchical model, combined with hidden Markov processes with four states for modeling spatial dependence, to detect genomic sites with epigenetic changes from two-sample experiments with paired internal control. One attractive feature of the proposed method is that the four states of the hidden Markov process have well-defined biological meanings and allow us to directly call the change patterns based on the corresponding posterior probabilities. In contrast, none of existing methods can offer this advantage. In addition, the proposed method offers great power in statistical inference by spatial smoothing (via hidden Markov modeling) and information pooling (via hierarchical modeling). Both simulation studies and real data analysis in a cocaine addiction study illustrate the reliability and success of this method.
Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation.
Abstract
Aberrant signaling through the class I phosphatidylinositol 3-kinase (PI3K)-Akt axis is frequent in human cancer. Here, we show that Beclin 1, an essential autophagy and tumor suppressor protein, is a target of the protein kinase Akt. Expression of a Beclin 1 mutant resistant to Akt-mediated phosphorylation increased autophagy, reduced anchorage-independent growth, and inhibited Akt-driven tumorigenesis. Akt-mediated phosphorylation of Beclin 1 enhanced its interactions with 14-3-3 and vimentin intermediate filament proteins, and vimentin depletion increased autophagy and inhibited Akt-driven transformation. Thus, Akt-mediated phosphorylation of Beclin 1 functions in autophagy inhibition, oncogenesis, and the formation of an autophagy-inhibitory Beclin 1/14-3-3/vimentin intermediate filament complex. These findings have broad implications for understanding the role of Akt signaling and intermediate filament proteins in autophagy and cancer.
The starvation hormone, fibroblast growth factor-21, extends lifespan in mice.
Abstract
Fibroblast growth factor-21 (FGF21) is a hormone secreted by the liver during fasting that elicits diverse aspects of the adaptive starvation response. Among its effects, FGF21 induces hepatic fatty acid oxidation and ketogenesis, increases insulin sensitivity, blocks somatic growth and causes bone loss. Here we show that transgenic overexpression of FGF21 markedly extends lifespan in mice without reducing food intake or affecting markers of NAD+ metabolism or AMP kinase and mTOR signaling. Transcriptomic analysis suggests that FGF21 acts primarily by blunting the growth hormone/insulin-like growth factor-1 signaling pathway in liver. These findings raise the possibility that FGF21 can be used to extend lifespan in other species.DOI:http://dx.doi.org/10.7554/eLife.00065.001.
Serum granulocyte colony-stimulating factor and Alzheimer's disease.
Abstract
Background
Granulocyte colony-stimulating factor (G-CSF) promotes the survival and function of neutrophils. G-CSF is also a neurotrophic factor, increasing neuroplasticity and suppressing apoptosis.
Methods
We analyzed G-CSF levels in 197 patients with probable Alzheimer's disease (AD) and 203 cognitively normal controls (NCs) from a longitudinal study by the Texas Alzheimer's Research and Care Consortium (TARCC). Data were analyzed by regression with adjustment for age, education, gender and APOE4 status.
Results
Serum G-CSF was significantly lower in AD patients than in NCs (β = -0.073; p = 0.008). However, among AD patients, higher serum G-CSF was significantly associated with increased disease severity, as indicated by lower Mini-Mental State Examination scores (β = -0.178; p = 0.014) and higher scores on the global Clinical Dementia Rating (CDR) scale (β = 0.170; p = 0.018) and CDR Sum of Boxes (β = 0.157; p = 0.035).
Conclusion
G-CSF appears to have a complex relationship with AD pathogenesis and may reflect different pathophysiologic processes at different illness stages.
Cell-free formation of RNA granules: bound RNAs identify features and components of cellular assemblies.
Abstract
Cellular granules lacking boundary membranes harbor RNAs and their associated proteins and play diverse roles controlling the timing and location of protein synthesis. Formation of such granules was emulated by treatment of mouse brain extracts and human cell lysates with a biotinylated isoxazole (b-isox) chemical. Deep sequencing of the associated RNAs revealed an enrichment for mRNAs known to be recruited to neuronal granules used for dendritic transport and localized translation at synapses. Precipitated mRNAs contain extended 3' UTR sequences and an enrichment in binding sites for known granule-associated proteins. Hydrogels composed of the low complexity (LC) sequence domain of FUS recruited and retained the same mRNAs as were selectively precipitated by the b-isox chemical. Phosphorylation of the LC domain of FUS prevented hydrogel retention, offering a conceptual means of dynamic, signal-dependent control of RNA granule assembly.
Slug, a unique androgen-regulated transcription factor, coordinates androgen receptor to facilitate castration resistance in prostate cancer.
Abstract
Prostate cancer (PCa) becomes lethal when cancer cells develop into castration-resistant PCa (CRPC). Androgen receptor (AR) gene mutation, altered AR regulation, or overexpression of AR often found in CRPC is believed to become one of the key factors to the lethal phenotype. Here we identify Slug, a member of the Snail family of zinc-finger transcription factors associated with cancer metastasis, as a unique androgen-responsive gene in PCa cells. In addition, the presence of constitutively active AR can induce Slug expression in a ligand-independent manner. Slug overexpression will increase AR protein expression and form a complex with AR. In addition, Slug appears to be a novel coactivator to enhance AR transcriptional activities and AR-mediated cell growth with or without androgen. In vivo, elevated Slug expression provides a growth advantage for PCa cells in androgen-deprived conditions. Most importantly, these observations were validated by several data sets from tissue microarrays. Overall, there is a reciprocal regulation between Slug and AR not only in transcriptional regulation but also in protein bioactivity, and Slug-AR complex plays an important role in accelerating the androgen-independent outgrowth of CRPC.
Effect of hypoxia and hyperoxia on cerebral blood flow, blood oxygenation, and oxidative metabolism.
Abstract
Characterizing the effect of oxygen (O(2)) modulation on the brain may provide a better understanding of several clinically relevant problems, including acute mountain sickness and hyperoxic therapy in patients with traumatic brain injury or ischemia. Quantifying the O(2) effects on brain metabolism is also critical when using this physiologic maneuver to calibrate functional magnetic resonance imaging (fMRI) signals. Although intuitively crucial, the question of whether the brain's metabolic rate depends on the amount of O(2) available has not been addressed in detail previously. This can be largely attributed to the scarcity and complexity of measurement techniques. Recently, we have developed an MR method that provides a noninvasive (devoid of exogenous agents), rapid (<5 minutes), and reliable (coefficient of variant, CoV <3%) measurement of the global cerebral metabolic rate of O(2) (CMRO(2)). In the present study, we evaluated metabolic and vascular responses to manipulation of the fraction of inspired O(2) (FiO(2)). Hypoxia with 14% FiO(2) was found to increase both CMRO(2) (5.0±2.0%, N=16, P=0.02) and cerebral blood flow (CBF) (9.8±2.3%, P<0.001). However, hyperoxia decreased CMRO(2) by 10.3±1.5% (P<0.001) and 16.9±2.7% (P<0.001) for FiO(2) of 50% and 98%, respectively. The CBF showed minimal changes with hyperoxia. Our results suggest that modulation of inspired O(2) alters brain metabolism in a dose-dependent manner.
Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis.
Abstract
Exercise has beneficial effects on human health, including protection against metabolic disorders such as diabetes. However, the cellular mechanisms underlying these effects are incompletely understood. The lysosomal degradation pathway, autophagy, is an intracellular recycling system that functions during basal conditions in organelle and protein quality control. During stress, increased levels of autophagy permit cells to adapt to changing nutritional and energy demands through protein catabolism. Moreover, in animal models, autophagy protects against diseases such as cancer, neurodegenerative disorders, infections, inflammatory diseases, ageing and insulin resistance. Here we show that acute exercise induces autophagy in skeletal and cardiac muscle of fed mice. To investigate the role of exercise-mediated autophagy in vivo, we generated mutant mice that show normal levels of basal autophagy but are deficient in stimulus (exercise- or starvation)-induced autophagy. These mice (termed BCL2 AAA mice) contain knock-in mutations in BCL2 phosphorylation sites (Thr69Ala, Ser70Ala and Ser84Ala) that prevent stimulus-induced disruption of the BCL2-beclin-1 complex and autophagy activation. BCL2 AAA mice show decreased endurance and altered glucose metabolism during acute exercise, as well as impaired chronic exercise-mediated protection against high-fat-diet-induced glucose intolerance. Thus, exercise induces autophagy, BCL2 is a crucial regulator of exercise- (and starvation)-induced autophagy in vivo, and autophagy induction may contribute to the beneficial metabolic effects of exercise.
Comparing statistical methods for constructing large scale gene networks.
Abstract
The gene regulatory network (GRN) reveals the regulatory relationships among genes and can provide a systematic understanding of molecular mechanisms underlying biological processes. The importance of computer simulations in understanding cellular processes is now widely accepted; a variety of algorithms have been developed to study these biological networks. The goal of this study is to provide a comprehensive evaluation and a practical guide to aid in choosing statistical methods for constructing large scale GRNs. Using both simulation studies and a real application in E. coli data, we compare different methods in terms of sensitivity and specificity in identifying the true connections and the hub genes, the ease of use, and computational speed. Our results show that these algorithms performed reasonably well, and each method has its own advantages: (1) GeneNet, WGCNA (Weighted Correlation Network Analysis), and ARACNE (Algorithm for the Reconstruction of Accurate Cellular Networks) performed well in constructing the global network structure; (2) GeneNet and SPACE (Sparse PArtial Correlation Estimation) performed well in identifying a few connections with high specificity.
Probe mapping across multiple microarray platforms.
Abstract
Access to gene expression data has become increasingly common in recent years; however, analysis has become more difficult as it is often desirable to integrate data from different platforms. Probe mapping across microarray platforms is the first and most crucial step for data integration. In this article, we systematically review and compare different approaches to map probes across seven platforms from different vendors: U95A, U133A and U133 Plus 2.0 from Affymetrix, Inc.; HT-12 v1, HT-12v2 and HT-12v3 from Illumina, Inc.; and 4112A from Agilent, Inc. We use a unique data set, which contains 56 lung cancer cell line samples-each of which has been measured by two different microarray platforms-to evaluate the consistency of expression measurement across platforms using different approaches. Based on the evaluation from the empirical data set, the BLAST alignment of the probe sequences to a recent revision of the Transcriptome generated better results than using annotations provided by Vendors or from Bioconductor's Annotate package. However, a combination of all three methods (deemed the 'Consensus Annotation') yielded the most consistent expression measurement across platforms. To facilitate data integration across microarray platforms for the research community, we develop a user-friendly web-based tool, an API and an R package to map data across different microarray platforms from Affymetrix, Illumina and Agilent. Information on all three can be found at http://qbrc.swmed.edu/software/probemapper/.
Development of methods for quantitative comparison of pooled shRNAs by mass sequencing.
Abstract
Pooled short-hairpin RNA (shRNA) library screening is a powerful tool for identifying a set of genes in biological pathways that require stable expression to produce a desired phenotype. Massive parallel sequencing of half-hairpins has proven highly variable and has not given satisfactory results concerning the relative abundance of different shRNAs before and after selection. Here, the authors describe a method for quantitative comparison of half-hairpins from pooled shRNAs in the mir30-based pGIPZ vector that is analyzed by massive parallel sequencing. Introducing a multiplexing code and refining the sample preparation scheme resulted in the predicted ability to detect twofold enrichments. These improvements should permit half-hairpin sequencing to analyze either dropout screens or selective pooled shRNA screens of limited stringency to analyze phenotypes not accessible in transient experiments.
Distinctive disruption patterns of white matter tracts in Alzheimer's disease with full diffusion tensor characterization.
Abstract
To characterize the white matter structural changes at the tract level and tract group level, comprehensive analysis with 4 metrics derived from diffusion tensor imaging (DTI), fractional anisotropy (FA), mean diffusivity (MD), axial diffusivity (AxD) and radial diffusivity (RD), was conducted. Tract groups, namely limbic, commissural, association, and projection tracts, include white matter tracts of similar functions. Diffusion tensor imaging data were acquired from 61 subjects (26 Alzheimer's disease [AD], 11 subjects with amnestic mild cognitive impairment [aMCI], and 24 age-matched controls). An atlas-based approach was used to survey 30 major cerebral white matter tracts with the measurements of FA, MD, AxD, and RD. Regional cortical atrophy and cognitive functions of AD patients were also measured to correlate with the structural changes of white matter. Synchronized structural changes of cingulum bundle and fornix, both of which are part of limbic tract group, were revealed. Widespread yet distinctive structural changes were found in limbic, commissural, association, and projection tract groups between control and AD subjects. Specifically, FA, MD, and RD of limbic tracts, FA, MD, AxD, and RD of commissural tracts, MD, AxD, and RD of association tracts, and MD and AxD of projection tracts are significantly different between AD patients and control subjects. In contrast, the comparison between aMCI and control subjects shows disruption only in the limbic and commissural tract groups of aMCI subjects. MD values of all tract groups of AD patients are significantly correlated to cognitive functions. Difference between AD and control and that between aMCI and control indicates a progression pattern of white matter disruption from limbic and commissural tract group to other tract groups. High correlation between FA, MD, and RD measurements from limbic tracts and cortical atrophy suggests the disruption of the limbic tract group is caused by the neuronal damage.
A blood-based screening tool for Alzheimer's disease that spans serum and plasma: findings from TARC and ADNI.
Abstract
Context
There is no rapid and cost effective tool that can be implemented as a front-line screening tool for Alzheimer's disease (AD) at the population level.
Objective
To generate and cross-validate a blood-based screener for AD that yields acceptable accuracy across both serum and plasma.
Design, Setting, Participants
Analysis of serum biomarker proteins were conducted on 197 Alzheimer's disease (AD) participants and 199 control participants from the Texas Alzheimer's Research Consortium (TARC) with further analysis conducted on plasma proteins from 112 AD and 52 control participants from the Alzheimer's Disease Neuroimaging Initiative (ADNI). The full algorithm was derived from a biomarker risk score, clinical lab (glucose, triglycerides, total cholesterol, homocysteine), and demographic (age, gender, education, APOE*E4 status) data.
Major Outcome Measures
Alzheimer's disease.
Results
11 proteins met our criteria and were utilized for the biomarker risk score. The random forest (RF) biomarker risk score from the TARC serum samples (training set) yielded adequate accuracy in the ADNI plasma sample (training set) (AUC = 0.70, sensitivity (SN) = 0.54 and specificity (SP) = 0.78), which was below that obtained from ADNI cerebral spinal fluid (CSF) analyses (t-tau/Aβ ratio AUC = 0.92). However, the full algorithm yielded excellent accuracy (AUC = 0.88, SN = 0.75, and SP = 0.91). The likelihood ratio of having AD based on a positive test finding (LR+) = 7.03 (SE = 1.17; 95% CI = 4.49-14.47), the likelihood ratio of not having AD based on the algorithm (LR-) = 3.55 (SE = 1.15; 2.22-5.71), and the odds ratio of AD were calculated in the ADNI cohort (OR) = 28.70 (1.55; 95% CI = 11.86-69.47).
Conclusions
It is possible to create a blood-based screening algorithm that works across both serum and plasma that provides a comparable screening accuracy to that obtained from CSF analyses.
Image-based genome-wide siRNA screen identifies selective autophagy factors.
Abstract
Selective autophagy involves the recognition and targeting of specific cargo, such as damaged organelles, misfolded proteins, or invading pathogens for lysosomal destruction. Yeast genetic screens have identified proteins required for different forms of selective autophagy, including cytoplasm-to-vacuole targeting, pexophagy and mitophagy, and mammalian genetic screens have identified proteins required for autophagy regulation. However, there have been no systematic approaches to identify molecular determinants of selective autophagy in mammalian cells. Here, to identify mammalian genes required for selective autophagy, we performed a high-content, image-based, genome-wide small interfering RNA screen to detect genes required for the colocalization of Sindbis virus capsid protein with autophagolysosomes. We identified 141 candidate genes required for viral autophagy, which were enriched for cellular pathways related to messenger RNA processing, interferon signalling, vesicle trafficking, cytoskeletal motor function and metabolism. Ninety-six of these genes were also required for Parkin-mediated mitophagy, indicating that common molecular determinants may be involved in autophagic targeting of viral nucleocapsids and autophagic targeting of damaged mitochondria. Murine embryonic fibroblasts lacking one of these gene products, the C2-domain containing protein, SMURF1, are deficient in the autophagosomal targeting of Sindbis and herpes simplex viruses and in the clearance of damaged mitochondria. Moreover, SMURF1-deficient mice accumulate damaged mitochondria in the heart, brain and liver. Thus, our study identifies candidate determinants of selective autophagy, and defines SMURF1 as a newly recognized mediator of both viral autophagy and mitophagy.
Robust gene expression signature from formalin-fixed paraffin-embedded samples predicts prognosis of non-small-cell lung cancer patients.
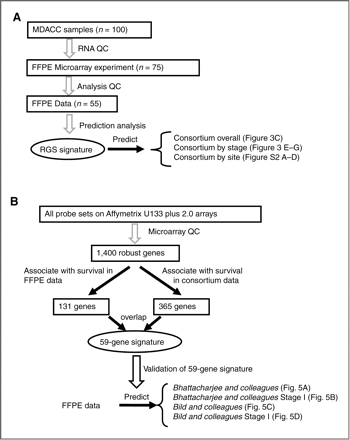
Abstract
Purpose
The requirement of frozen tissues for microarray experiments limits the clinical usage of genome-wide expression profiling by using microarray technology. The goal of this study is to test the feasibility of developing lung cancer prognosis gene signatures by using genome-wide expression profiling of formalin-fixed paraffin-embedded (FFPE) samples, which are widely available and provide a valuable rich source for studying the association of molecular changes in cancer and associated clinical outcomes.
Experimental Design
We randomly selected 100 Non-Small-Cell lung cancer (NSCLC) FFPE samples with annotated clinical information from the UT-Lung SPORE Tissue Bank. We microdissected tumor area from FFPE specimens and used Affymetrix U133 plus 2.0 arrays to attain gene expression data. After strict quality control and analysis procedures, a supervised principal component analysis was used to develop a robust prognosis signature for NSCLC. Three independent published microarray datasets were used to validate the prognosis model.
Results
This study showed that the robust gene signature derived from genome-wide expression profiling of FFPE samples is strongly associated with lung cancer clinical outcomes and can be used to refine the prognosis for stage I lung cancer patients, and the prognostic signature is independent of clinical variables. This signature was validated in several independent studies and was refined to a 59-gene lung cancer prognosis signature.
Conclusion
We conclude that genome-wide profiling of FFPE lung cancer samples can identify a set of genes whose expression level provides prognostic information across different platforms and studies, which will allow its application in clinical settings.
A blood-based algorithm for the detection of Alzheimer's disease.
Abstract
Background
We previously created a serum-based algorithm that yielded excellent diagnostic accuracy in Alzheimer's disease. The current project was designed to refine that algorithm by reducing the number of serum proteins and by including clinical labs. The link between the biomarker risk score and neuropsychological performance was also examined.
Methods
Serum-protein multiplex biomarker data from 197 patients diagnosed with Alzheimer's disease and 203 cognitively normal controls from the Texas Alzheimer's Research Consortium were analyzed. The 30 markers identified as the most important from our initial analyses and clinical labs were utilized to create the algorithm.
Results
The 30-protein risk score yielded a sensitivity, specificity, and AUC of 0.88, 0.82, and 0.91, respectively. When combined with demographic data and clinical labs, the algorithm yielded a sensitivity, specificity, and AUC of 0.89, 0.85, and 0.94, respectively. In linear regression models, the biomarker risk score was most strongly related to neuropsychological tests of language and memory.
Conclusions
Our previously published diagnostic algorithm can be restricted to only 30 serum proteins and still retain excellent diagnostic accuracy. Additionally, the revised biomarker risk score is significantly related to neuropsychological test performance.
Modeling Three-Dimensional Chromosome Structures Using Gene Expression Data.
Abstract
Introduction
Recent genomic studies have shown that significant chromosomal spatial correlation exists in gene expression of many organisms. Interestingly, coexpression has been observed among genes separated by a fixed interval in specific regions of a chromosome chain, which is likely caused by three-dimensional (3D) chromosome folding structures. Modeling such spatial correlation explicitly may lead to essential understandings of 3D chromosome structures and their roles in transcriptional regulation. In this paper, we explore chromosomal spatial correlation induced by 3D chromosome structures, and propose a hierarchical Bayesian method based on helical structures to formally model and incorporate the correlation into the analysis of gene expression microarray data. It is the first study to quantify and infer 3D chromosome structures in vivo using expression microarrays. Simulation studies show computing feasibility of the proposed method and that, under the assumption of helical chromosome structures, it can lead to precise estimation of structural parameters and gene expression levels. Real data applications demonstrate an intriguing biological phenomenon that functionally associated genes, which are far apart along the chromosome chain, are brought into physical proximity by chromosomal folding in 3D space to facilitate their coexpression. It leads to important biological insight into relationship between chromosome structure and function.
A novel approach to DNA copy number data segmentation.
Abstract
DNA copy number (DCN) is the number of copies of DNA at a region of a genome. The alterations of DCN are highly associated with the development of different tumors. Recently, microarray technologies are being employed to detect DCN changes at many loci at the same time in tumor samples. The resulting DCN data are often very noisy, and the tumor sample is often contaminated by normal cells. The goal of computational analysis of array-based DCN data is to infer the underlying DCNs from raw DCN data. Previous methods for this task do not model the tumor/normal cell mixture ratio explicitly and they cannot output segments with DCN annotations. We developed a novel model-based method using the minimum description length (MDL) principle for DCN data segmentation. Our new method can output underlying DCN for each chromosomal segment, and at the same time, infer the underlying tumor proportion in the test samples. Empirical results show that our method achieves better accuracies on average as compared to three previous methods, namely Circular Binary Segmentation, Hidden Markov Model and Ultrasome.
White matter cerebral blood flow is inversely correlated with structural and functional connectivity in the human brain.
Abstract
White matter provides anatomic connections among brain regions and has received increasing attention in understanding brain intrinsic networks and neurological disorders. Despite significant progresses made in characterizing the white matter's structural properties using post-mortem techniques and in vivo diffusion-tensor-imaging (DTI) methods, its physiology remains poorly understood. In the present study, cerebral blood flow (CBF) of the white matter was investigated on a fiber tract-specific basis using MRI (n=10, 25-33 years old). It was found that CBF in the white matter varied considerably, up to a factor of two between fiber groups. Furthermore, a paradoxically inverse correlation was observed between white matter CBF and structural and functional connectivities (P<0.001). Fiber tracts that had a higher CBF tended to have a lower fractional anisotropy in water diffusion, and the gray matter terminals connected to the tract also tended to have a lower temporal synchrony in resting-state BOLD signal fluctuation. These findings suggest a clear association between white matter perfusion and gray matter activity, but the nature of this relationship requires further investigations given that they are negatively, rather than positively, correlated.
Nuclear receptor expression defines a set of prognostic biomarkers for lung cancer.
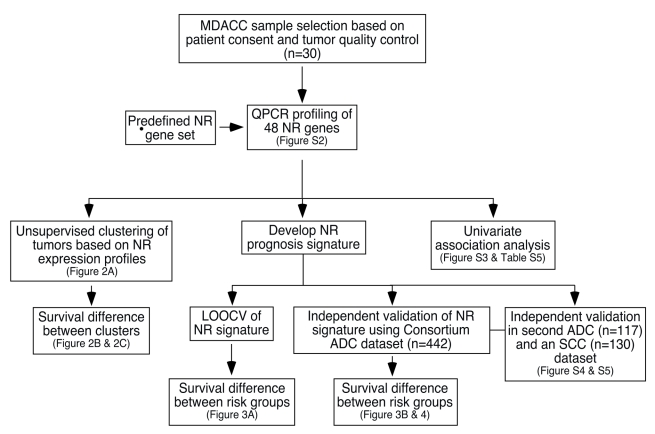
Abstract
Background
The identification of prognostic tumor biomarkers that also would have potential as therapeutic targets, particularly in patients with early stage disease, has been a long sought-after goal in the management and treatment of lung cancer. The nuclear receptor (NR) superfamily, which is composed of 48 transcription factors that govern complex physiologic and pathophysiologic processes, could represent a unique subset of these biomarkers. In fact, many members of this family are the targets of already identified selective receptor modulators, providing a direct link between individual tumor NR quantitation and selection of therapy. The goal of this study, which begins this overall strategy, was to investigate the association between mRNA expression of the NR superfamily and the clinical outcome for patients with lung cancer, and to test whether a tumor NR gene signature provided useful information (over available clinical data) for patients with lung cancer.
Methods and Findings
Using quantitative real-time PCR to study NR expression in 30 microdissected non-small-cell lung cancers (NSCLCs) and their pair-matched normal lung epithelium, we found great variability in NR expression among patients' tumor and non-involved lung epithelium, found a strong association between NR expression and clinical outcome, and identified an NR gene signature from both normal and tumor tissues that predicted patient survival time and disease recurrence. The NR signature derived from the initial 30 NSCLC samples was validated in two independent microarray datasets derived from 442 and 117 resected lung adenocarcinomas. The NR gene signature was also validated in 130 squamous cell carcinomas. The prognostic signature in tumors could be distilled to expression of two NRs, short heterodimer partner and progesterone receptor, as single gene predictors of NSCLC patient survival time, including for patients with stage I disease. Of equal interest, the studies of microdissected histologically normal epithelium and matched tumors identified expression in normal (but not tumor) epithelium of NGFIB3 and mineralocorticoid receptor as single gene predictors of good prognosis.
Conclusion
NR expression is strongly associated with clinical outcomes for patients with lung cancer, and this expression profile provides a unique prognostic signature for lung cancer patient survival time, particularly for those with early stage disease. This study highlights the potential use of NRs as a rational set of therapeutically tractable genes as theragnostic biomarkers, and specifically identifies short heterodimer partner and progesterone receptor in tumors, and NGFIB3 and MR in non-neoplastic lung epithelium, for future detailed translational study in lung cancer. Please see later in the article for the Editors' Summary.
A serum protein-based algorithm for the detection of Alzheimer disease.
Abstract
Objective
To develop an algorithm that separates patients with Alzheimer disease (AD) from controls.
Design
Longitudinal case-control study.
Setting
The Texas Alzheimer's Research Consortium project. Patients We analyzed serum protein-based multiplex biomarker data from 197 patients diagnosed with AD and 203 controls. Main Outcome Measure The total sample was randomized equally into training and test sets and random forest methods were applied to the training set to create a biomarker risk score.
Results
The biomarker risk score had a sensitivity and specificity of 0.80 and 0.91, respectively, and an area under the curve of 0.91 in detecting AD. When age, sex, education, and APOE status were added to the algorithm, the sensitivity, specificity, and area under the curve were 0.94, 0.84, and 0.95, respectively.
Conclusions
These initial data suggest that serum protein-based biomarkers can be combined with clinical information to accurately classify AD. A disproportionate number of inflammatory and vascular markers were weighted most heavily in the analyses. Additionally, these markers consistently distinguished cases from controls in significant analysis of microarray, logistic regression, and Wilcoxon analyses, suggesting the existence of an inflammatory-related endophenotype of AD that may provide targeted therapeutic opportunities for this subset of patients.
Induced Sézary syndrome PBMCs poorly express immune response genes up-regulated in stimulated memory T cells.
Abstract
Background
Dysfunctions in memory T cells contribute to various inflammatory autoimmune diseases and neoplasms. We hypothesize that investigating the differences of genetic profiles between resting and activated naïve and memory T cells may provide insight into the characterization of abnormal memory T cells in diseases, such as Sézary syndrome (SS), a neoplasm composed of CD4(+) CD45RO(+) cells.
Objective
We determined genes distinctively expressed between resting and activated naive and memory cells. Levels of up-regulated genes in resting and activated memory cells were measured in SS PBMCs, which were largely comprised of CD4(+) CD45RO(+) cells, to quantitatively assess how different Sézary cells were from memory cells.
Methods
We compared gene expression profiles using high-density oligo-microarrays between resting and activated naïve and memory CD4(+) T cells. Differentially expressed genes were confirmed by qRT-PCR and immunoblotting. Levels of genes up-regulated in activated and resting memory T cells were determined in SS PBMCs by qRT-PCR.
Results
Activated memory cells expressed greater numbers of immune-mediated genes involved in effector function compared to naïve cells in our microarray analysis and qRT-PCR. Nine out of 14 genes with enhanced levels in activated memory cells had reduced levels in SS PBMCs (p<0.05).
Conclusions
Activation of memory and naïve CD4(+) T cells revealed a diverging gap in gene expression between these subsets, with memory cells expressing immune-related genes important for effector function. Many of these genes were markedly depressed in SS patients, implying Sézary cells are markedly impaired in mounting immune responses compared to memory cells.
Dnmt3a regulates emotional behavior and spine plasticity in the nucleus accumbens.
Abstract
Despite abundant expression of DNA methyltransferases (Dnmts) in brain, the regulation and behavioral role of DNA methylation remain poorly understood. We found that Dnmt3a expression was regulated in mouse nucleus accumbens (NAc) by chronic cocaine use and chronic social defeat stress. Moreover, NAc-specific manipulations that block DNA methylation potentiated cocaine reward and exerted antidepressant-like effects, whereas NAc-specific Dnmt3a overexpression attenuated cocaine reward and was pro-depressant. On a cellular level, we found that chronic cocaine use selectively increased thin dendritic spines on NAc neurons and that DNA methylation was both necessary and sufficient to mediate these effects. These data establish the importance of Dnmt3a in the NAc in regulating cellular and behavioral plasticity to emotional stimuli.
Improving fMRI sensitivity by normalization of basal physiologic state.
Abstract
Introduction
Chemotherapy prolongs survival without substantially impairing quality of life for medically fit patients with advanced non-small cell lung cancer (NSCLC), but population-based studies have shown that only 20 to 30% of these patients receive chemotherapy. These earlier studies have relied on Medicare-linked Surveillance, Epidemiology, and End Results (SEER) data, thus excluding the 30 to 35% of lung cancer patients younger than 65 years. Therefore, we determined the use of chemotherapy in a contemporary, diverse NSCLC population encompassing all patient ages.
Methods
We performed a retrospective analysis of patients diagnosed with stage IV NSCLC from 2000 to 2007 at the University of Texas Southwestern Medical Center. Demographic, treatment, and outcome data were obtained from hospital tumor registries. The association between these variables was assessed using univariate analysis and multivariate logistic regression.
Results
In all, 718 patients met criteria for analysis. Mean age was 60 years, 58% were men, and 45% were white. Three hundred fifty-three patients (49%) received chemotherapy. In univariate analysis, receipt of chemotherapy was associated with age (53% of patients younger than 65 years versus 41% of patients aged 65 years and older; p = 0.003) and insurance type (p < 0.001). In a multivariate model, age and insurance type remained associated with receipt of chemotherapy. For individuals receiving chemotherapy, median survival was 9.2 months, compared with 2.3 months for untreated patients (p < 0.001).
Conclusion
In a contemporary population representing the full age range of patients with advanced NSCLC, chemotherapy was administered to approximately half of all patients-more than twice the rate reported in some earlier studies. Patient age and insurance type are associated with receipt of chemotherapy.
A Bayesian approach to joint modeling of protein-DNA binding, gene expression and sequence data.
Abstract
The genome-wide DNA-protein-binding data, DNA sequence data and gene expression data represent complementary means to deciphering global and local transcriptional regulatory circuits. Combining these different types of data can not only improve the statistical power, but also provide a more comprehensive picture of gene regulation. In this paper, we propose a novel statistical model to augment protein-DNA-binding data with gene expression and DNA sequence data when available. We specify a hierarchical Bayes model and use Markov chain Monte Carlo simulations to draw inferences. Both simulation studies and an analysis of an experimental data set show that the proposed joint modeling method can significantly improve the specificity and sensitivity of identifying target genes as compared with conventional approaches relying on a single data source.
Imipramine treatment and resiliency exhibit similar chromatin regulation in the mouse nucleus accumbens in depression models.
Abstract
Although it is a widely studied psychiatric syndrome, major depressive disorder remains a poorly understood illness, especially with regard to the disconnect between treatment initiation and the delayed onset of clinical improvement. We have recently validated chronic social defeat stress in mice as a model in which a depression-like phenotype is reversed by chronic, but not acute, antidepressant administration. Here, we use chromatin immunoprecipitation (ChIP)-chip assays--ChIP followed by genome wide promoter array analyses--to study the effects of chronic defeat stress on chromatin regulation in the mouse nucleus accumbens (NAc), a key brain reward region implicated in depression. Our results demonstrate that chronic defeat stress causes widespread and long-lasting changes in gene regulation, including alterations in repressive histone methylation and in phospho-CREB (cAMP response element-binding protein) binding, in the NAc. We then show similarities and differences in this regulation to that observed in another mouse model of depression, prolonged adult social isolation. In the social defeat model, we observed further that many of the stress-induced changes in gene expression are reversed by chronic imipramine treatment, and that resilient mice-those resistant to the deleterious effects of defeat stress-show patterns of chromatin regulation in the NAc that overlap dramatically with those seen with imipramine treatment. These findings provide new insight into the molecular basis of depression-like symptoms and the mechanisms by which antidepressants exert their delayed clinical efficacy. They also raise the novel idea that certain individuals resistant to stress may naturally mount antidepressant-like adaptations in response to chronic stress.
Genome-wide analysis of chromatin regulation by cocaine reveals a role for sirtuins.
Abstract
Changes in gene expression contribute to the long-lasting regulation of the brain's reward circuitry seen in drug addiction; however, the specific genes regulated and the transcriptional mechanisms underlying such regulation remain poorly understood. Here, we used chromatin immunoprecipitation coupled with promoter microarray analysis to characterize genome-wide chromatin changes in the mouse nucleus accumbens, a crucial brain reward region, after repeated cocaine administration. Our findings reveal several interesting principles of gene regulation by cocaine and of the role of DeltaFosB and CREB, two prominent cocaine-induced transcription factors, in this brain region. The findings also provide comprehensive insight into the molecular pathways regulated by cocaine-including a new role for sirtuins (Sirt1 and Sirt2)-which are induced in the nucleus accumbens by cocaine and, in turn, dramatically enhance the behavioral effects of the drug.
The receptor interacting protein 1 inhibits p53 induction through NF-kappaB activation and confers a worse prognosis in glioblastoma.
Abstract
Nuclear factor-kappaB (NF-kappaB) activation may play an important role in the pathogenesis of cancer and also in resistance to treatment. Inactivation of the p53 tumor suppressor is a key component of the multistep evolution of most cancers. Links between the NF-kappaB and p53 pathways are under intense investigation. In this study, we show that the receptor interacting protein 1 (RIP1), a central component of the NF-kappaB signaling network, negatively regulates p53 tumor suppressor signaling. Loss of RIP1 from cells results in augmented induction of p53 in response to DNA damage, whereas increased RIP1 level leads to a complete shutdown of DNA damage-induced p53 induction by enhancing levels of cellular mdm2. The key signal generated by RIP1 to up-regulate mdm2 and inhibit p53 is activation of NF-kappaB. The clinical implication of this finding is shown in glioblastoma, the most common primary malignant brain tumor in adults. We show that RIP1 is commonly overexpressed in glioblastoma, but not in grades II and III glioma, and increased expression of RIP1 confers a worse prognosis in glioblastoma. Importantly, RIP1 levels correlate strongly with mdm2 levels in glioblastoma. Our results show a key interaction between the NF-kappaB and p53 pathways that may have implications for the targeted treatment of glioblastoma.
On the assessment of cerebrovascular reactivity using hypercapnia BOLD MRI.
Abstract
Cerebrovascular reactivity (CVR) reflects the capacity of blood vessels to dilate and is an important marker for brain vascular reserve. It may provide a useful addition to the traditional baseline blood flow measurement when assessing vascular factors in brain disorders. Blood-oxygenation-level-dependent MRI under CO(2) inhalation offers a non-invasive and quantitative means to estimate CVR in humans. In this study, we investigated several important methodological aspects of this technique with the goal of optimizing the experimental and data processing strategies for clinical use. Comparing 4 min of 5% CO(2) inhalation (less comfortable) to a 1 min inhalation (more comfortable) duration, it was found that the CVR values were 0.31 +/- 0.05%/mmHg (N = 11) and 0.31 +/- 0.08%/mmHg (N = 9), respectively, showing no significant differences between the two breathing paradigms. Therefore, the 1 min paradigm is recommended for future application studies for patient comfort and tolerability. Furthermore, we have found that end-tidal CO(2) recording was useful for accurate quantification of CVR because it provided both timing and amplitude information regarding the input function to the brain vascular system, which can be subject-dependent. Finally, we show that inter-subject variations in CVR are of physiologic origin and affect the whole brain in a similar fashion. Based on this, it is proposed that relative CVR (normalized against the CVR of the whole brain or a reference tissue) may be a more sensitive biomarker than absolute CVR in clinical applications as it minimizes inter-subject variations. With these technological optimizations, CVR mapping may become a useful method for studies of neurological and psychiatric diseases.
Characterization of a novel ghrelin cell reporter mouse.
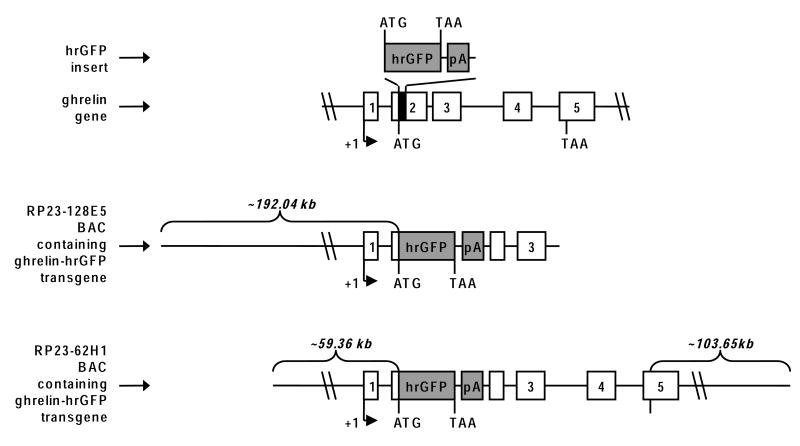
Abstract
Ghrelin is a hormone that influences many physiological processes and behaviors, such as food intake, insulin and growth hormone release, and a coordinated response to chronic stress. However, little is known about the molecular pathways governing ghrelin release and ghrelin cell function. To better study ghrelin cell physiology, we have generated several transgenic mouse lines expressing humanized Renilla reniformis green fluorescent protein (hrGFP) under the control of the mouse ghrelin promoter. hrGFP expression was especially abundant in the gastric oxyntic mucosa, in a pattern mirroring that of ghrelin immunoreactivity and ghrelin mRNA. hrGFP expression also was observed in the duodenum, but not in the brain, pancreatic islet, or testis. In addition, we used fluorescent activated cell sorting (FACS) to collect and partially characterize highly enriched populations of gastric ghrelin cells. We suggest that these novel ghrelin-hrGFP transgenic mice will serve as useful tools to better understand ghrelin cell physiology.
Improved detection of differentially expressed genes through incorporation of gene locations.
Abstract
In determining differential expression in cDNA microarray experiments, the expression level of an individual gene is usually assumed to be independent of the expression levels of other genes, but many recent studies have shown that a gene's expression level tends to be similar to that of its neighbors on a chromosome, and differentially expressed (DE) genes are likely to form clusters of similar transcriptional activity along the chromosome. When modeled as a one-dimensional spatial series, the expression level of genes on the same chromosome frequently exhibit significant spatial correlation, reflecting spatial patterns in transcription. By modeling these spatial correlations, we can obtain improved estimates of transcript levels. Here, we demonstrate the existence of spatial correlations in transcriptional activity in the Escherichia coli (E. coli) chromosome across more than 50 experimental conditions. Based on this finding, we propose a hierarchical Bayesian model that borrows information from neighboring genes to improve the estimation of the expression level of a given gene and hence the detection of DE genes. Furthermore, we extend the model to account for the circular structure of E. coli chromosome and the intergenetic distance between gene neighbors. The simulation studies and analysis of real data examples in E. coli and yeast Saccharomyces cerevisiae show that the proposed method outperforms the commonly used significant analysis of microarray (SAM) t-statistic in detecting DE genes.
Cocaine regulates MEF2 to control synaptic and behavioral plasticity.
Abstract
Repeated exposure to cocaine causes sensitized behavioral responses and increased dendritic spines on medium spiny neurons of the nucleus accumbens (NAc). We find that cocaine regulates myocyte enhancer factor 2 (MEF2) transcription factors to control these two processes in vivo. Cocaine suppresses striatal MEF2 activity in part through a mechanism involving cAMP, the regulator of calmodulin signaling (RCS), and calcineurin. We show that reducing MEF2 activity in the NAc in vivo is required for the cocaine-induced increases in dendritic spine density. Surprisingly, we find that increasing MEF2 activity in the NAc, which blocks the cocaine-induced increase in dendritic spine density, enhances sensitized behavioral responses to cocaine. Together, our findings implicate MEF2 as a key regulator of structural synapse plasticity and sensitized responses to cocaine and suggest that reducing MEF2 activity (and increasing spine density) in NAc may be a compensatory mechanism to limit long-lasting maladaptive behavioral responses to cocaine.
Enhanced identification and biological validation of differential gene expression via Illumina whole-genome expression arrays through the use of the model-based background correction methodology.
Abstract
Despite the tremendous growth of microarray usage in scientific studies, there is a lack of standards for background correction methodologies, especially in single-color microarray platforms. Traditional background subtraction methods often generate negative signals and thus cause large amounts of data loss. Hence, some researchers prefer to avoid background corrections, which typically result in the underestimation of differential expression. Here, by utilizing nonspecific negative control features integrated into Illumina whole genome expression arrays, we have developed a method of model-based background correction for BeadArrays (MBCB). We compared the MBCB with a method adapted from the Affymetrix robust multi-array analysis algorithm and with no background subtraction, using a mouse acute myeloid leukemia (AML) dataset. We demonstrated that differential expression ratios obtained by using the MBCB had the best correlation with quantitative RT-PCR. MBCB also achieved better sensitivity in detecting differentially expressed genes with biological significance. For example, we demonstrated that the differential regulation of Tnfr2, Ikk and NF-kappaB, the death receptor pathway, in the AML samples, could only be detected by using data after MBCB implementation. We conclude that MBCB is a robust background correction method that will lead to more precise determination of gene expression and better biological interpretation of Illumina BeadArray data.
TAR DNA-binding protein 43 immunohistochemistry reveals extensive neuritic pathology in FTLD-U: a midwest-southwest consortium for FTLD study.
Abstract
TAR DNA-binding protein 43 (TDP-43) is a major component of the inclusions in frontotemporal lobar degeneration with ubiquitinated inclusions (FTLD-U). We studied TDP-43 pathology in the hippocampus and frontal cortex of autopsy brains from patients with FTLD-U (n = 68), dementia lacking distinctive histopathology (n = 4), other neurodegenerative diseases (n = 23), and controls (n = 12) using a sensitive immunohistochemistry protocol. Marked enhancement of staining of TDP-43-positive dystrophic neurites (DNs) was obtained, and we observed 2 previously unrecognized pathologic patterns (i.e. frequent long DNs in the CA1 region and frequent dot-like DNs in the neocortical layer 2) in 39% and 15% of the FTLD-U cases, respectively. Frequent long DNs, but not dot-like DNs, were significantly associated with progranulin mutations. Based on this evaluation, 4 FTLD-U cases showed no TDP-43 pathology and were reclassified as "FTLD-U, non-TDP-43 proteinopathy," and 3 cases of dementia lacking distinctive histopathology were reclassified as FTLD-U. Of the cases with other neurodegenerative diseases, 43% showed TDP-43 pathology in the hippocampus, but only 4% showed TDP-43 pathology in the frontal cortex. No TDP-43 pathology was seen in controls. These results indicate that the sensitivity of the TDP-43 immunohistochemistry method affects both the extent and type of abnormalities detected. Moreover, assessment of abnormalities in both the hippocampus and frontal cortex may be diagnostically important in FTLD-U.
Distinct roles of adenylyl cyclases 1 and 8 in opiate dependence: behavioral, electrophysiological, and molecular studies.
Abstract
Background
Opiate dependence is a result of adaptive changes in signal transduction networks in several brain regions. Noradrenergic neurons of the locus coeruleus (LC) have provided a useful model system in which to understand the molecular basis of these adaptive changes. One of most robust signaling adaptations to repeated morphine exposure in this brain region is upregulation of adenylyl cyclase (AC) activity. Earlier work revealed the selective induction of two calmodulin-dependent AC isoforms, AC1 and AC8, after chronic morphine, but their role in opiate dependence has remained unknown.
Methods
Whole cell recordings from LC slices, behavioral paradigms for dependence, and gene array technology have been used to dissect the role of AC1 and AC8 in chronic morphine responses.
Results
Both AC1 and AC8 knockout mice exhibit reduced opiate dependence on the basis of attenuated withdrawal; however, partially distinct withdrawal symptoms were affected in the two lines. Loss of AC1 or AC8 also attenuated the electrophysiological effects of morphine on LC neurons: knockout of either cyclase attenuated the chronic morphine-induced enhancement of baseline firing rates as well as of regulation of neuronal firing by forskolin (an activator of ACs). The DNA microarray analysis revealed that both AC1 and AC8 affect gene regulation in the LC by chronic morphine and, in addition to common genes, each cyclase influences the expression of a distinct subset of genes.
Conclusions
Together, these findings provide fundamentally new insight into the molecular and cellular basis of opiate