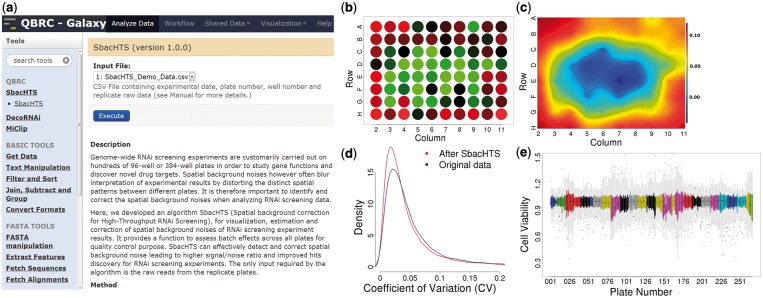
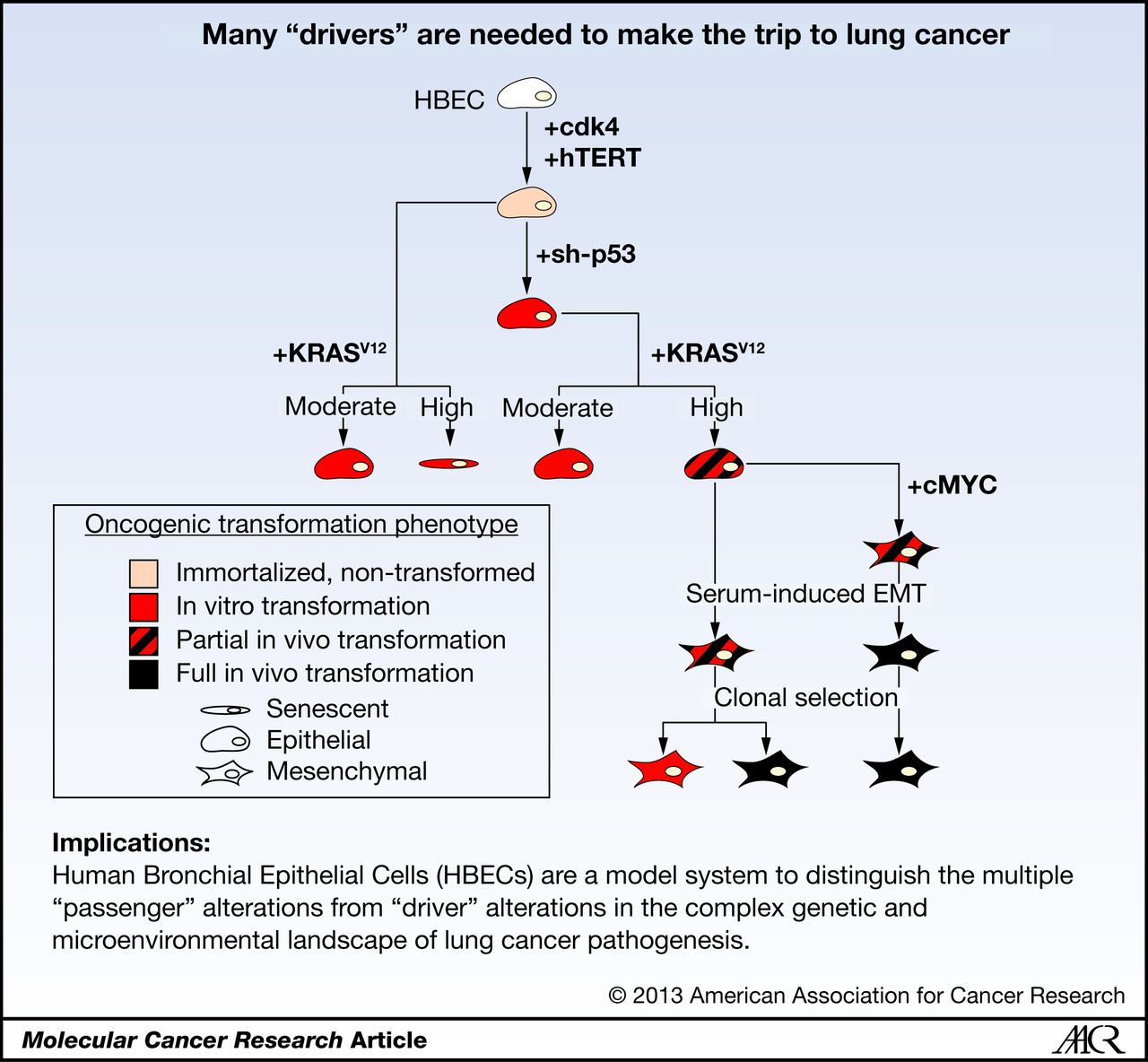
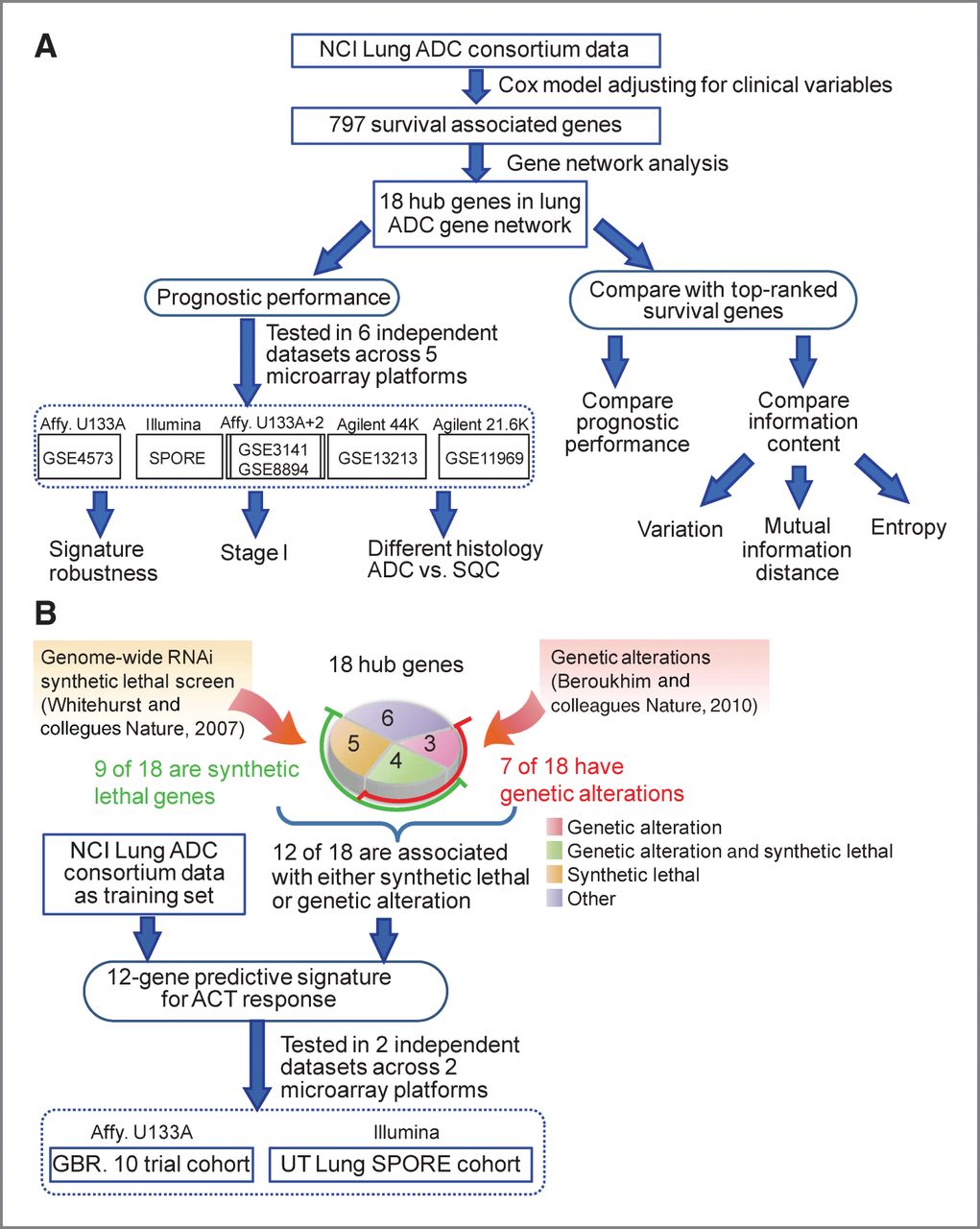
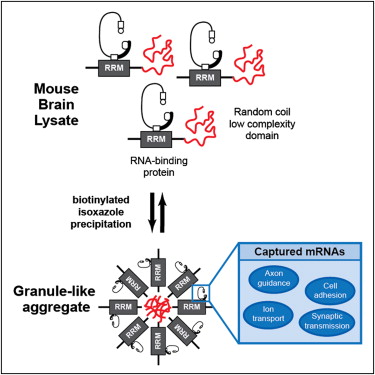
Welcome to Xie Lab
Short Biography
I am Professor and the Founding Chair of the Department of Health Data Science and Biostatistics in the Peter O’Donnell Jr. School of Public Health and the Associate Dean for Data Sciences at the University of Texas Southwestern (UTSW). I also served as the Founding Director of the Quantitative Biomedical Research Center (QBRC) and led the Data Science for Precision Health Initiative (DAPHI). Over the past 20 years at UTSW, I have built a nationally recognized research program that integrates biostatistics, artificial intelligence (AI), and clinical informatics to advance precision medicine.
Read More
Education
Ph.D. in Biostatistics, 2006
University of Minnesota-Twin Cities
M.S. in Biostatistics, 2002
University of Minnesota-Twin Cities
M.S in Epidemiology, 2000
Peking Union Medical College
BMedSc 1997
Peking University Health Science Center
Research Summary
Biomarker Discovery and Clinical Outcome Prediction
We have developed computational models to predict patient outcomes, allowing clinicians to tailor treatment plans for individual patients ...
Learn moreMethods for High-dimension Data and Integrative Analysis
We have developed bioinformatics tools, computational algorithms and statistical methodologies for the processing and analysis of high dimensional ...
Learn moreStatistical Learning and Prediction Model
My lab has extensive experience in develop predictive models in biomedical research. Our team has won several highly competitive international computational challenges ...
Learn moreRNA Regulation
Over the past couple of decades, a surge of discoveries have revealed RNA regulation as a central player in cellular processes. RNAs are regulated by RNA-binding proteins (RBPs) at all post-transcriptional stages. ...
Learn moreResearch Interests
Biomarker discovery and validation
Genomic data analysis and data integration
Medical informatics
Clinical trial design
Lung cancer
Predicting the future for people with lung cancer
Nature Medicine. 2008 Aug;14(8):812-3. PubMed PMID: 18685594;
PubMed Central PMCID: PMC2833359.
Xie Y, Minna JD. Predicting the future for people with lung cancer.
A lung cancer molecular prognostic test ready for prime time
Lancet, 379, 785-787.
Xie, Y., and Minna, J. D.
A 12-Gene Set Predicts Survival Benefits from Adjuvant Chemotherapy in Non-Small-Cell Lung Cancer Patients
Clin Cancer Res; January 28, 2013;
doi:10.1158/1078-0432.CCR-12-2321. (*Corresponding Author)
Tang H, Xiao G, Behrens C, Schiller J, Allen J, Chow CW, Suraokar M, Corvalan A, Mao
JH, White M, Wistuba II, Minna J, Xie, Y.

Using artificial intelligence to personalize lung cancer treatment
UTSW research combining artificial intelligence with traditional pathology analysis holds potential for quickly creating a personalized attack plan for cancer patients when speed is essential: as non-small cell lung cancers spread. This approach identified lung cancers that are most likely to respond to one common treatment versus those that might benefit from a different approach.

Congratulations to Dr. Yang Xie appointed inaugural Associate Deans
"I feel honored to serve as the inaugural Associate Dean of Data Sciences because data science is playing increasing roles in all aspects of medical research," Dr. Xie said. "How to analyze, interpret, and utilize this data to understand biology to help with patient care and public health – that has become more and more important."